The genome sequence of the flounced rustic, Luperina testacea (Denis & Schiffermüller, 1775)
Gavin R. Broad, Nicholas W VanKuren, Francesco Cicconardi, Susan McEvoy

TL;DR
This paper presents the genome sequence of the flounced rustic moth, including a detailed assembly of its chromosomes and mitochondrial DNA.
Contribution
The study provides a high-quality genome assembly for Luperina testacea, including chromosomal scaffolding and the mitochondrial genome.
Findings
The genome assembly is 601 megabases in size.
99.98% of the assembly is organized into 31 chromosomal pseudomolecules, including the Z sex chromosome.
The mitochondrial genome is 15.3 kilobases long.
Abstract
We present a genome assembly from an individual male Luperina testacea (the flounced rustic; Arthropoda; Insecta; Lepidoptera; Noctuidae). The genome sequence is 601 megabases in span. The majority of the assembly (99.98%) is scaffolded into 31 chromosomal pseudomolecules, with the Z sex chromosome assembled. The mitochondrial genome was also assembled, and is 15.3 kilobases in length.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1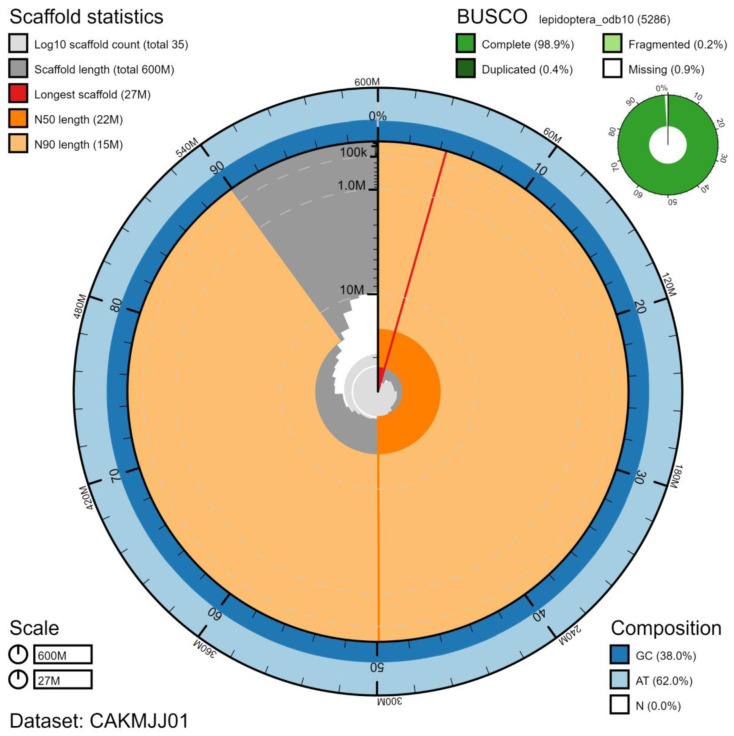 Figure 2
Figure 2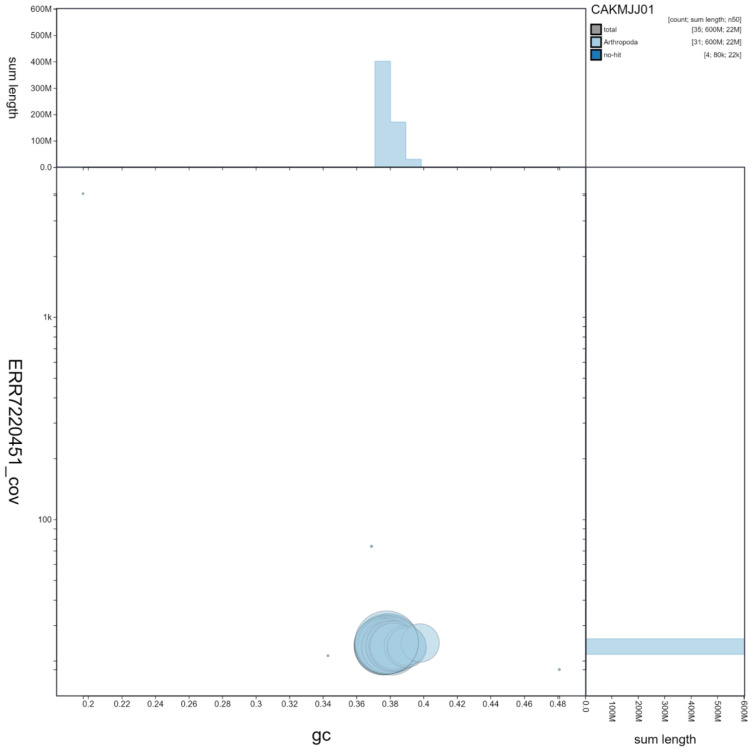 Figure 3
Figure 3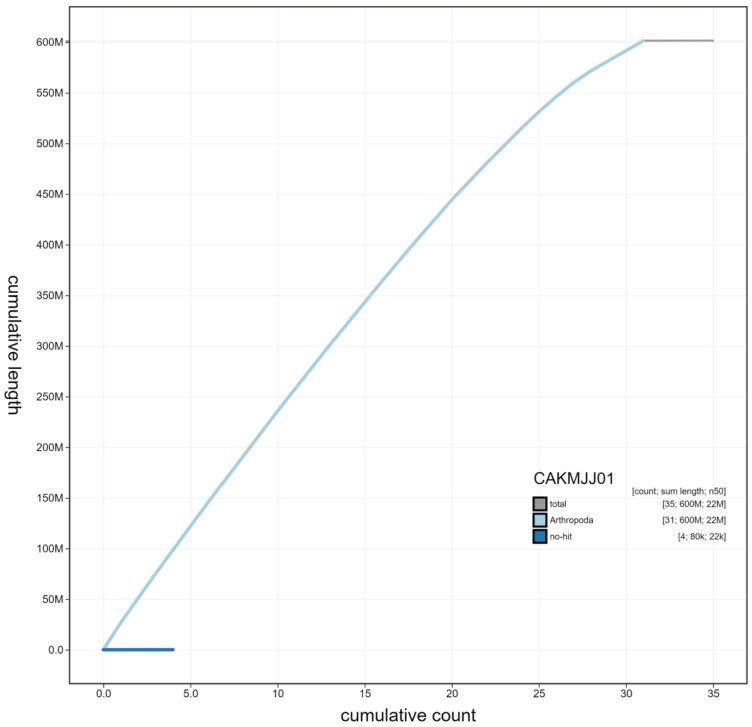 Figure 4
Figure 4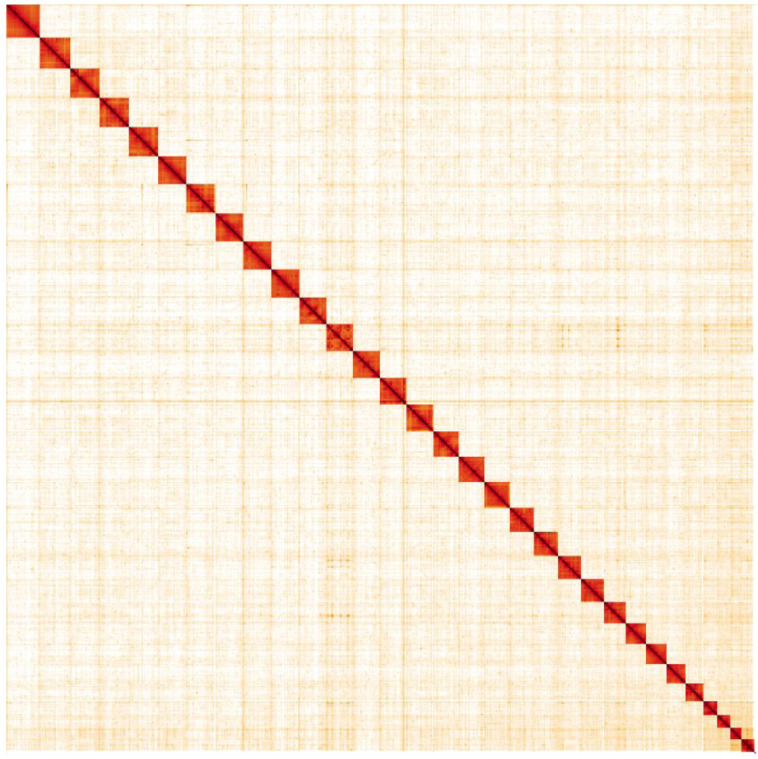 Figure 5
Figure 5|
| |
|---|---|
| Assembly identifier | ilLupTest1.1 |
| Species |
|
| Specimen | ilLupTest1 |
| NCBI taxonomy ID | NCBI:txid988174 |
| BioProject | PRJEB48331 |
| BioSample ID | SAMEA8534287 |
| Isolate information | Male, thorax (genome
|
|
| |
| PacificBiosciences SEQUEL II | ERR7221643 |
| 10X Genomics Illumina | ERR7220448-ERR7220451 |
| Hi-C Illumina | ERR7220452 |
|
| |
| Assembly accession | GCA_927399505.1 |
| Accession of alternate haplotype | GCA_927399495.1 |
| Span (Mb) | 601 |
| Number of contigs | 68 |
| Contig N50 length (Mb) | 17.1 |
| Number of scaffolds | 34 |
| Scaffold N50 length (Mb) | 21.6 |
| Longest scaffold (Mb) | 24.7 |
| BUSCO
| C:98.9%[S:98.6%,D:0.4%],
|
| INSDC accession | Chromosome | Size (Mb) | GC% |
|---|---|---|---|
| 1 | 24.71 | 37.8 | |
| 2 | 23.72 | 38.0 | |
| 3 | 23.38 | 37.9 | |
| 4 | 23.35 | 37.7 | |
| 5 | 23.31 | 38.0 | |
| 6 | 22.77 | 37.6 | |
| 7 | 22.63 | 37.6 | |
| 8 | 22.60 | 38.0 | |
| 9 | 22.54 | 37.7 | |
| 10 | 21.93 | 37.6 | |
| 11 | 21.69 | 38.1 | |
| 12 | 21.61 | 37.6 | |
| 13 | 21.13 | 37.8 | |
| 14 | 20.98 | 38.0 | |
| 15 | 20.97 | 37.8 | |
| 16 | 20.67 | 37.7 | |
| 17 | 20.09 | 38.1 | |
| 18 | 19.70 | 38.2 | |
| 19 | 19.66 | 37.9 | |
| 20 | 18.24 | 37.9 | |
| 21 | 18.03 | 38.1 | |
| 22 | 17.37 | 38.4 | |
| 23 | 17.07 | 38.1 | |
| 24 | 16.30 | 38.2 | |
| 25 | 15.06 | 38.2 | |
| 26 | 13.89 | 38.2 | |
| 27 | 11.55 | 38.9 | |
| 28 | 10.17 | 39.0 | |
| 29 | 9.84 | 39.0 | |
| 30 | 9.66 | 39.8 | |
| Z | 26.81 | 37.8 | |
| MT | 0.02 | 20.1 | |
| - | Unplaced | 0.06 | 39.8 |
| Software tool | Version | Source |
|---|---|---|
| Hifiasm | 0.15.3 |
|
| purge_dups | 1.2.3 |
|
| SALSA | 2.2 |
|
| longranger align | 2.2.2 |
|
| freebayes | 1.3.1-17-
|
|
| MitoHiFi | 2.0 |
|
| HiGlass | 1.11.6 |
|
| PretextView | 0.2.x |
|
| BlobToolKit | 3.0.5 |
|
- —Wellcome Trust
- —Wellcome Trust
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic diversity and population structure · Plant and animal studies · Poxvirus research and outbreaks
Species taxonomy
Eukaryota; Metazoa; Ecdysozoa; Arthropoda; Hexapoda; Insecta; Pterygota; Neoptera; Endopterygota; Lepidoptera; Glossata; Ditrysia; Noctuoidea; Noctuidae; Noctuinae; Apameini; Luperina; Luperina testacea (Denis & Schiffermüller, 1775) (NCBI:txid988002).
Background
Across much of north-west and central Europe, Luperina testacea, the flounced rustic, is a common moth of late summer and autumn. Widespread in Britain, Luperina testacea becomes much less frequent in Scotland. Eggs are laid at the bases of various grasses and the smooth, rather unpigmented larvae feed on the roots and lower stems, feeding slowly through the winter. Adults do not feed, are regular at light (predominantly males) and can be one of the most abundantly trapped moths from late July to early October, especially in open coastal areas or calcareous grasslands ( Henwood et al., 2020; Waring et al., 2003). Abundance in southern Sweden is positively associated with soil disturbance ( Tyler, 2020).
Adults are variable in appearance, particularly in the tone of the forewing. The hind wings are always strikingly pale, the fore wings usually have a distinct dark bar in the centre and a broad, paler subterminal area.
Genome sequence report
The genome was sequenced from one male L. testacea ( Figure 1) collected from Hever Castle, England, UK (latitude 51.1884, longitude 0.1198). A total of 26-fold coverage in Pacific Biosciences single-molecule long reads and 76-fold coverage in 10X Genomics read clouds were generated. Primary assembly contigs were scaffolded with chromosome conformation Hi-C data. Manual assembly curation corrected 24 missing/misjoins, reducing the assembly size by 0.01% and the scaffold number by 33.33%, and increasing the scaffold N50 by 4.57%.
Image of the Luperina testacea (ilLupTest1) specimen taken prior to preservation and processing.
The final assembly has a total length of 601 Mb in 34 sequence scaffolds with a scaffold N50 of 21.6 Mb ( Table 1). The majority of the assembly sequence (99.98%) was assigned to 31 chromosomal-level scaffolds, representing 30 autosomes (numbered by sequence length), and the Z sex chromosome ( Figure 2– Figure 5; Table 2). The assembly has a BUSCO v5.2.2 ( Manni et al., 2021) completeness of 98.9% (single 98.6%, duplicated 0.4%) using the lepidoptera_odb10 reference set. While not fully phased, the assembly deposited is of one haplotype. Contigs corresponding to the second haplotype have also been deposited.
Table 1.: Genome data for Luperina testacea, ilLupTest1.1.
Genome assembly of Luperina testacea, ilLupTest1.1: metrics.The BlobToolKit Snailplot shows N50 metrics and BUSCO gene completeness. The main plot is divided into 1,000 size-ordered bins around the circumference with each bin representing 0.1% of the 601,512,407 bp assembly. The distribution of chromosome lengths is shown in dark grey with the plot radius scaled to the longest chromosome present in the assembly (26,814,763 bp, shown in red). Orange and pale-orange arcs show the N50 and N90 chromosome lengths (21,613,865 and 15,062,659 bp), respectively. The pale grey spiral shows the cumulative chromosome count on a log scale with white scale lines showing successive orders of magnitude. The blue and pale-blue area around the outside of the plot shows the distribution of GC, AT and N percentages in the same bins as the inner plot. A summary of complete, fragmented, duplicated and missing BUSCO genes in the lepidoptera_odb10 set is shown in the top right. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/ilLupTest1.1/dataset/CAKMJJ01/snail.
Genome assembly of Luperina testacea, ilLupTest1.1: GC coverage.BlobToolKit GC-coverage plot. Scaffolds are coloured by phylum. Circles are sized in proportion to scaffold length Histograms show the distribution of scaffold length sum along each axis. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/ilLupTest1.1/dataset/CAKMJJ01/blob.
Genome assembly of Luperina testacea, ilLupTest1.1: cumulative sequence.BlobToolKit cumulative sequence plot. The grey line shows cumulative length for all scaffolds. Coloured lines show cumulative lengths of scaffolds assigned to each phylum using the buscogenes taxrule. An interactive version of this figure is available at https://blobtoolkit.genomehubs.org/view/ilLupTest1.1/dataset/CAKMJJ01/cumulative.
Genome assembly of Luperina testacea, ilLupTest1.1: Hi-C contact map.Hi-C contact map of the ilNotZicz1.1 assembly, visualised in HiGlass. Chromosomes are shown in order of size from left to right and top to bottom. An interactive version of this map is available here.
Table 2.: Chromosomal pseudomolecules in the genome assembly of Luperina testacea, ilLupTest1.1.
Methods
Sample acquisition and DNA extraction
A single male L. testacea (ilLupTest1) was collected from Hever Castle, England, UK (latitude 51.1884, longitude 0.1198) by Gavin Broad, Natural History Museum, using a light trap in grassland near a lake. The sample was identified by the same individual, and preserved in liquid nitrogen.
DNA was extracted at the Tree of Life laboratory, Wellcome Sanger Institute. The ilLupTest1 sample was weighed and dissected on dry ice with tissue set aside for Hi-C sequencing. Thorax tissue was cryogenically disrupted to a fine powder using a Covaris cryoPREP Automated Dry Pulveriser, receiving multiple impacts. Fragment size analysis of 0.01–0.5 ng of DNA was then performed using an Agilent FemtoPulse. High molecular weight (HMW) DNA was extracted using the Qiagen MagAttract HMW DNA extraction kit. Low molecular weight DNA was removed from a 200-ng aliquot of extracted DNA using 0.8X AMpure XP purification kit prior to 10X Chromium sequencing; a minimum of 50 ng DNA was submitted for 10X sequencing. HMW DNA was sheared into an average fragment size between 12–20 kb in a Megaruptor 3 system with speed setting 30. Sheared DNA was purified by solid-phase reversible immobilisation using AMPure PB beads with a 1.8X ratio of beads to sample to remove the shorter fragments and concentrate the DNA sample. The concentration of the sheared and purified DNA was assessed using a Nanodrop spectrophotometer and Qubit Fluorometer and Qubit dsDNA High Sensitivity Assay kit. Fragment size distribution was evaluated by running the sample on the FemtoPulse system.
Sequencing
Pacific Biosciences HiFi circular consensus and 10X Genomics Chromium read cloud sequencing libraries were constructed according to the manufacturers’ instructions. Sequencing was performed by the Scientific Operations core at the Wellcome Sanger Institute on Pacific Biosciences SEQUEL II (HiFi) and Illumina NovaSeq 6000 (10X) instruments. Hi-C data were generated from head tissue using the Arima Hi-C+ kit and sequenced on NovaSeq 6000.
Genome assembly
Assembly was carried out with Hifiasm ( Cheng et al., 2021); haplotypic duplication was identified and removed with purge_dups ( Guan et al., 2020). One round of polishing was performed by aligning 10X Genomics read data to the assembly with longranger align, calling variants with freebayes ( Garrison & Marth, 2012). The assembly was then scaffolded with Hi-C data ( Rao et al., 2014) using SALSA2 ( Ghurye et al., 2019). The assembly was checked for contamination as described previously ( Howe et al., 2021). Manual curation ( Howe et al., 2021) was performed using HiGlass ( Kerpedjiev et al., 2018) and Pretext. The mitochondrial genome was assembled using MitoHiFi ( Uliano-Silva et al., 2021), which performs annotation using MitoFinder ( Allio et al., 2020). The genome was analysed and BUSCO scores generated within the BlobToolKit environment ( Challis et al., 2020). Table 3 contains a list of all software tool versions used, where appropriate.
Ethics/compliance issues
The materials that have contributed to this genome note have been supplied by a Darwin Tree of Life Partner. The submission of materials by a Darwin Tree of Life Partner is subject to the Darwin Tree of Life Project Sampling Code of Practice. By agreeing with and signing up to the Sampling Code of Practice, the Darwin Tree of Life Partner agrees they will meet the legal and ethical requirements and standards set out within this document in respect of all samples acquired for, and supplied to, the Darwin Tree of Life Project. Each transfer of samples is further undertaken according to a Research Collaboration Agreement or Material Transfer Agreement entered into by the Darwin Tree of Life Partner, Genome Research Limited (operating as the Wellcome Sanger Institute), and in some circumstances other Darwin Tree of Life collaborators.
Data availability
European Nucleotide Archive: Luperina testacea. Accession number PRJEB48331; https://identifiers.org/ena.embl/PRJEB48331.
The genome sequence is released openly for reuse. The L. testacea genome sequencing initiative is part of the Darwin Tree of Life (DToL) project. All raw sequence data and the assembly have been deposited in INSDC databases. The genome will be annotated and presented through the Ensembl pipeline at the European Bioinformatics Institute. Raw data and assembly accession identifiers are reported in Table 1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Allio R Schomaker-Bastos A Romiguier J : Mito Finder: Efficient Automated Large-Scale Extraction of Mitogenomic Data in Target Enrichment Phylogenomics. Mol Ecol Resour. 2020;20(4):892–905. 10.1111/1755-0998.13160 32243090 PMC 7497042 · doi ↗ · pubmed ↗
- 2Challis R Richards E Rajan J : Blob Tool Kit-Interactive Quality Assessment of Genome Assemblies. G 3 (Bethesda). 2020;10(4):1361–1374. 10.1534/g 3.119.400908 32071071 PMC 7144090 · doi ↗ · pubmed ↗
- 3Cheng H Concepcion GT Feng X : Haplotype-Resolved de Novo Assembly Using Phased Assembly Graphs with Hifiasm. Nat Methods. 2021;18(2):170–75. 10.1038/s 41592-020-01056-5 33526886 PMC 7961889 · doi ↗ · pubmed ↗
- 4Garrison E Marth G : Haplotype-Based Variant Detection from Short-Read Sequencing. ar Xiv: 1207.3907.2012. 10.48550/ar Xiv.1207.3907 · doi ↗
- 5Ghurye J Rhie A Walenz BP : Integrating Hi-C Links with Assembly Graphs for Chromosome-Scale Assembly. P Lo S Comput Biol. 2019;15(8):e 1007273. 10.1371/journal.pcbi.1007273 31433799 PMC 6719893 · doi ↗ · pubmed ↗
- 6Guan D Mc Carthy SA Wood J : Identifying and Removing Haplotypic Duplication in Primary Genome Assemblies. Bioinformatics. 2020;36(9):2896–2898. 10.1093/bioinformatics/btaa 025 31971576 PMC 7203741 · doi ↗ · pubmed ↗
- 7Henwood B Sterling P Lewington R : Field Guide to the Caterpillars of Great Britain and Ireland. Bloomsbury Publishing. 2020. Reference Source
- 8Howe K Chow W Collins J : Significantly Improving the Quality of Genome Assemblies through Curation. Gigascience. 2021;10(1):giaa 153. 10.1093/gigascience/giaa 153 33420778 PMC 7794651 · doi ↗ · pubmed ↗