A Novel Pathogenic TUBA1A Variant in a Croatian Infant Is Linked to a Severe Tubulinopathy with Walker–Warburg-like Features
Akzam Saidin, Anet Papazovska Cherepnalkovski, Zeeshan Shaukat, Todor Arsov, Rashid Hussain, Ben J. Roberts, Marija Bucat, Klara Cogelja, Michael G. Ricos, Leanne M. Dibbens

TL;DR
A rare TUBA1A gene mutation in a Croatian infant caused severe neurological issues resembling Walker–Warburg Syndrome but with distinct features.
Contribution
A novel TUBA1A mutation is linked to a new tubulinopathy with macrocephaly and hypoplastic genitalia, not previously associated with the gene.
Findings
A novel de novo TUBA1A mutation (p.His283Arg) was identified in an infant with severe neurological features.
Clinical features included macrocephaly, hypoplastic genitalia, and hypotonia without eye anomalies.
The condition is distinct from Walker–Warburg Syndrome and represents a new tubulinopathy.
Abstract
Tubulinopathies are associated with malformations of cortical development but not Walker–Warburg Syndrome. Intensive monitoring of a Croatian infant presenting as Walker–Warburg Syndrome in utero began at 21 weeks due to increased growth of cerebral ventricles and foetal biparietal diameter. Monitoring continued until Caesarean delivery at 34 weeks where the infant was eutrophic. Clinical assessment of a progressive neurological disorder of unknown aetiology found a macrocephalic head and markedly hypoplastic genitalia with a micropenis. Neurological examination showed generalized hypotonia with very rare spontaneous movements, hypotonia-induced respiratory insufficiency and ventilator dependence, and generalized myoclonus intensifying during manipulation. With clinical features of hypotonia, lissencephaly, and brain malformations, Walker–Warburg Syndrome was suspected; however, eye…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
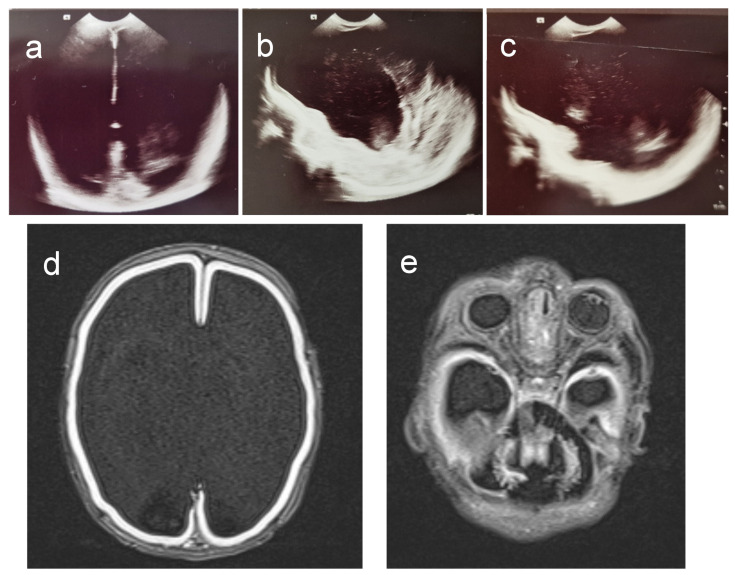 Figure 1
Figure 1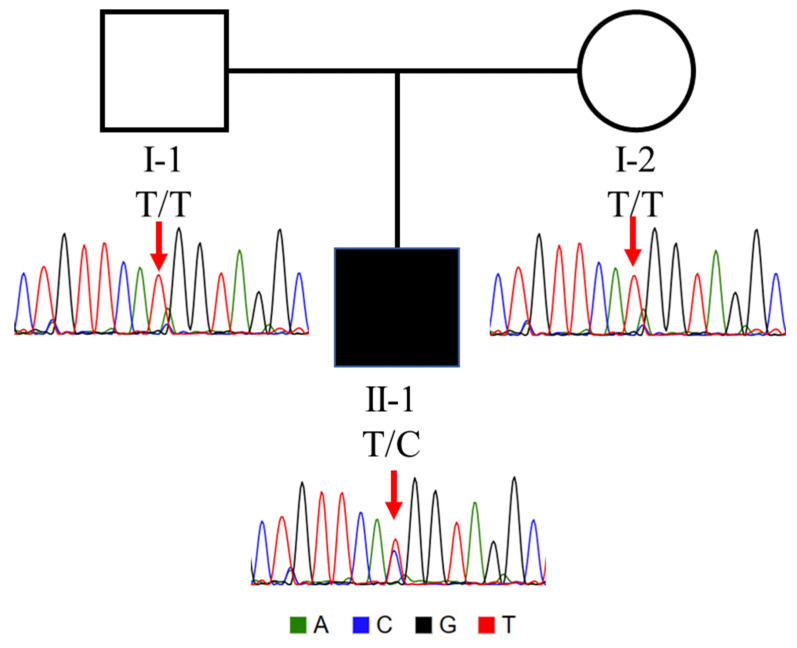 Figure 2
Figure 2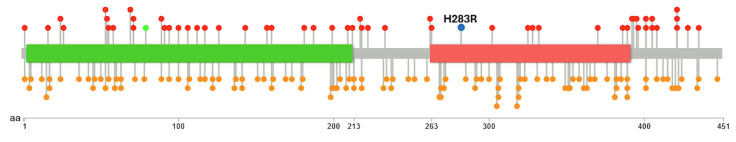 Figure 3
Figure 3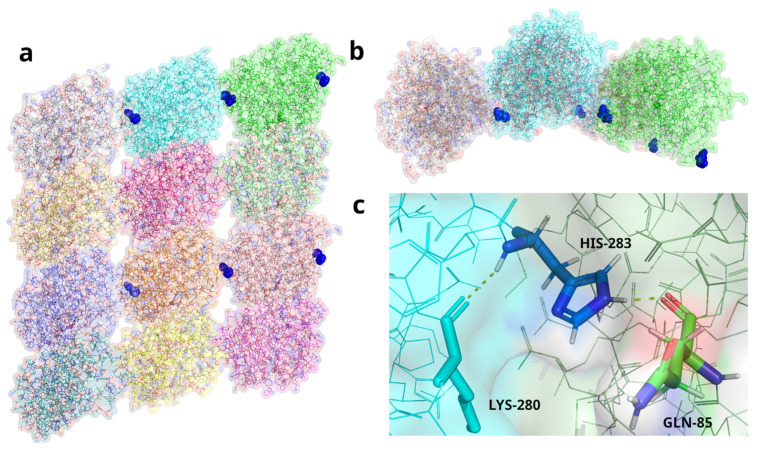 Figure 4
Figure 4- —University of South Australia
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRNA regulation and disease · Mitochondrial Function and Pathology · Infective Endocarditis Diagnosis and Management
1. Introduction
Tubulinopathies are a heterogeneous group of disorders caused by mutations in members of the tubulin superfamily of genes [1]. Among the five tubulin families, members from the α-α and β-β families encode tubulin proteins which form heterodimers and are two of the main components of microtubules [1]. Tubulins are involved in a range of cellular processes including intracellular transport, cell division, and neuronal migration [1]. While tubulinopathies have broad clinical presentations, many include cortical and subcortical brain malformations such as lissencephaly, polymicrogyria, and cortical dysplasia [1,2]. Other clinical features include microcephaly, global developmental delay, intellectual disability, and seizures [1]. TUBA1A mutations are the most common cause of tubulinopathies with neurological phenotypes, and to date, all identified pathogenic TUBA1A mutations appear to be autosomal dominant heterozygous loss of function mutations [1].
Walker–Warburg Syndrome (WWS) is a rare autosomal recessive disease where patients present with several comorbid phenotypes including brain malformations (both lissencephaly and cerebellar malformations), eye malformations, and congenital muscular dystrophy [3,4]. To date, the majority of genes associated with WWS, like O-Mannosyltransferase Gene (POMT1) [5] and Fukutin-related protein (FKRP) [6], have been implicated in causing defects in the glycosylation of proteins mainly of α-dystroglycan encoded by the DAG1 gene, which is also a cause of WWS [7]. As such, WWS is now considered by many to be, first and foremost, a congenital muscular dystrophy caused by α-dystroglycan glycosylation defects with associated brain and eye abnormalities [7].
We investigated an infant of Croatian origin that was born with severe developmental abnormalities of hypotonia and cobblestone lissencephaly type brain malformations indicative of Walker–Warburg Syndrome (WWS) but lacked the associated eye phenotypes. Clinical investigations revealed hydrocephalus and ventriculomegaly indicative of WWS, but the infant also had macrocephalic features, which are generally atypical of WWS. Genetic investigation of the affected infant and their parents by whole-exome trio sequencing revealed a novel de novo heterozygous TUBA1A mutation c.848A>G, p.His283Arg that, based on its predicted effects via in silico analysis and 3D protein modelling, was the most likely cause of the disease phenotypes. Based on the presence of the clinical features of hypertonia and brain malformations, Walker–Warburg Syndrome was initially suspected, but the absence of eye phenotypes and the identification of a mutation in the TUBA1A gene, which has not been shown to cause glycosylation defects or WWS, did not support a WWS diagnosis. This ultra rare and pathogenic mutation in the TUBA1A gene represents a new TUBA1A-associated tubulinopathy caused by a previously unpublished TUBA1A mutation and a phenotype that should not be conflated with canonical WWS.
2. Materials and Methods
2.1. Clinical Investigation
Clinical investigation of the infant was performed at the Clinical Centre Split, Croatia. Ultrasound of the brain was performed using a Siemens Healthcare d.o.o., Zagreb, Croatia, Healthineers Acuson NX3 Elite, ultrasound device with a convex transducer frequency of 7.3 MHz. Brain MRI was performed using a Siemens AG, Erlangen, Germany, Magnetom Symphony 1.5 Tesla device.
2.2. Genetic Investigation
Chromosomal analysis of a peripheral blood sample from the infant was conducted using Giemsa trypsin (GTG banding). Trio whole-exome sequencing (WES) was performed on genomic DNA extracted from peripheral blood from the patient and their biological parents. DNA libraries were prepared using SureSelect Human All Exon V7 (Agilent, Santa Clara, CA, USA) and sequenced on the Illumina NovaSeq platform (Illumina, San Diego, CA, USA). NovoAlign [8] was used for reads alignment to human reference genome (GRCh38), and the Genome Analysis Toolkit (GATK) HaplotypeCaller was used for joint variant calling [9]. Variant annotation was performed using SnpEff [10] and SnpSift [11] against dbNSFPv4 [12]. Slivar was used for variant prioritization with various inheritance models and to identify structural deletions in parent–child duos using non-transmission of alleles [13]. There was only a single protein-altering de novo sequence mutation identified, and this was classified according to ACMG guidelines using InterVar [14]. Bidirectional Sanger sequencing of genomic DNA confirmed the presence of the mutation in the patient and its absence in the parents.
A lollipop plot of non-synonymous mutations from clinvar was generated for the TUBA1A protein (UniProt: Q71U36) using the tool lollipops [15]. A total of 167 non-synonymous mutations were extracted from clinvar, comprising 54 pathogenic, 112 likely pathogenic, 1 likely benign, and 0 benign variants. In an effort to uncover genotype–phenotype relationships, we extracted only the pathogenic and likely pathogenic mutations that had accompanying phenotypic information (150 from clinvar of which a subset of 77 mutations is also reported in Tantry et al. (2023) [16]). We then focused on variants located in the tubulin C-terminal domain to compare the associated TUBA1A phenotypes.
2.3. Analysis of Mutation Location in the 3D Structure of Tubulin α-1A Chain Protein in a Complex with Tubulin β-III Protein
We performed protein visualization with the software PyMOL (v2.5.5) [17] on the published structure of recombinant neuronal human tubulin (Tubulin α-1A chain protein in a complex with Tubulin β-III protein), 5JCO [18], identifying the mutated residue of interest. The location of the Histidine (residue 283), which is mutated to Arginine, and its hydrogen bonds were marked/highlighted on the α-Tubulin monomers in the 3D tubulin structure.
3. Results
3.1. Clinical Investigations
The male patient was the second child from the second pregnancy of young non-consanguineous parents; the mother was 32 years of age and the father 34 at the time of birth. A healthy male sibling was 1.5 years old. The diagnosis of a neurological disorder was made prenatally at 21 weeks’ gestation when enhanced ventriculomegaly and rapidly increasing foetal biparietal diameter was detected during fetal ultrasound. The pregnancy was subsequently closely monitored. Serology for Toxoplasmosis, Rubella, CMV, HSV, and Syphilis were performed and excluded acute infection. Table 1 shows a summary of clinical features. The infant was delivered by elective caesarean section at 34 weeks’ gestation with a birth weight of 2.46 kg (61st percentile), birth length of 48 cm (73rd percentile), and a head circumference of 38.5 cm (>99th percentile) [19]. APGAR scores were 5 after 1 min (1 for each function) and improved to 6 (2 for pulse, 1 for the other functions) after 5 min. The infant was resuscitated using the T-piece resuscitator. After applying 5 initial sustained inflation breaths lasting 1 s each, a positive pressure ventilation was continued for 1 min that resulted in an increased heart rate to 120 beats per minute. However, spontaneous breathing was inefficient and superficial, so the child was immediately intubated at the beginning of the second minute of life and was mechanically ventilated thereafter.
Physical examination at birth found the infant to be eutrophic with a macrocephalic head. The sagittal suture was opened for 1.5 cm, and the large fontanel measured 5 × 6 cm while the small fontanel 1 × 2 cm. The abdomen was soft and the liver palpable at the right costal margin. Genitalia were extremely hypoplastic with barely indicated empty scrotum and a micropenis. Extremities appeared neat on the outside. Generalized hypotonia was observed with extremely rare spontaneous movements.
A brain ultrasound showed a medially positioned interhemispheric fissure. Cerebral hemispheres appeared dilated, filled with cerebrospinal fluid, and a thin mantle of the brain was visible on the brain convexity. The basal ganglia were not visible, nor were the choroid plexuses. The pons and cerebellum appeared hypoplastic (Figure 1a–c). Brain MRI showed an anomalous shape of all structures of the brainstem in the infratentorial area that morphologically shaped into the letter “Z”. The peduncles were extremely elongated and took on the shape of a “molar tooth sign”. The pons was flattened and folded ventrally. The vermis was not differentiated, and the cerebellar hemispheres were extremely hypoplastic. The fourth brain ventricle appeared voluminous and communicated with the mega cistern cerebellomedularis in terms of a Dandy Walker malformation/variant. Supratentorial, extremely hydrocephalically dilated lateral ventricles were visible, and the falx was differentiated in the ventral and dorsal parts. The cerebral hemispheres were reduced to a very narrow sheath of parenchyma, without the formation of sulci and gyri, suggesting a possible form of lissencephaly. Along the narrow parenchyma layer, marginally, in the presumed areas of the temporal horns, smaller, symmetrical crescent-shaped thickenings could be seen, which could correspond to lateralized and unconnected basal ganglia, i.e., rudimentary parts of the basal ganglia structures. In the sellar region, a very narrow Turkish saddle was observed. EEG activity showed pathological findings of diminished basic brain activity, suggestive of seizures. Echocardiography revealed patent atrial septal defect (ASD secundum) measuring 5 mm. The clinical presentation and complex congenital brain malformation (Figure 1d,e) indicated a Walker–Warburg Syndrome-like phenotype without eye anomalies.
The patient was intubated at birth due to respiratory insufficiency and was mechanically ventilated during the entire stay. Empiric antibiotic therapy was prescribed. He developed generalized myoclonus, mainly during manipulation. Phenobarbitone and then midazolam was prescribed continuously until the twelfth day that led to diminishing of the motor manifestations. Meconium discharge was supported with glycerin suppository application, and oral tube feeding was practiced. Diuresis was established from the beginning; however, he became anuric on the seventh day despite optimal fluid intake. Diuresis was maintained with a dopamine infusion.
On day 15, pleuropneumonia due to Staphylococcus warneri was detected and treated with meropenem and vancomycin, as well as by transfusion of erythrocyte concentrates. On day 43, transfusion of erythrocytes was repeated. The infant died at 2 months of age from respiratory insufficiency and cardiopulmonary arrest. Genetic analysis was undertaken to look for a cause of the fatal infantile neurological disorder.
3.2. Genetic Investigation
Chromosomal analysis of the infant showed a normal male karyotype of 46, XY. Whole-exome sequencing trio analysis of the patient and their biological parents was undertaken. We used slivar duo-del [13] to look for large deletions in the region of the exomes that arose de novo in the child. No large deletions were detected within the exome data.
Small nucleotide variant (SNV) analysis was prioritized according to the likely inheritance models of de novo, X-linked, and recessive mutations (including compound heterozygotes). In our exome sequence analysis, a single nonsynonymous de novo heterozygous mutation was identified, p.His283Arg in TUBA1A: Chr12 (GRCh38): g.49185518T>C: NM_006009.4: c.848A>G, NP_006000.2:, as the most likely cause. We also manually examined the exome data for mutations in known WWS genes, OrphaNet: https://www.orpha.net/en/disease/gene/list/899 (accessed on 27 July 2024), and no causative variants were found.
The TUBA1A H283R mutation is extremely rare, appearing in only a single entry in clinvar (VCV001701083.1; rs2121243281), deposited by a clinical sequencing centre and listed as “likely pathogenic” with a single phenotypic descriptor of “seizure”. This entry of the mutation remains unreported in the literature and there are no assertation criteria provided (clinvar-0 stars). We performed direct Sanger sequencing of genomic DNA, which confirmed the presence of the mutation in the affected infant and the absence in the unaffected parents (Figure 2), confirming that the mutation was de novo. Initial InterVar [14] ACMG classification prediction for the mutation was adjusted from likely pathogenic (PM1 + PM2 + PP2 + PP3) to pathogenic (PS2 + PM1 + PM2 + PP2 + PP3); PS2 criteria were added based on Sanger validation confirmation (Figure 2). The PM1 criteria were supported by the absence of non-synonymous benign variants, and more than 10 pathogenic or likely pathogenic reported variants within the surrounding region (Figure 3) [20]. Multiple in silico predictions predicted that the mutation is damaging; SIFT (0.0), Polyphen2 (HVAR 0.929), MutationTaster (0.999), GERP++ (5.1), and REVEL (0.907). The CADD score of 25 strongly suggested pathogenicity. The c.848A>G mutation is in exon 4 of TUBA1A and is not present in the ALFA, ExAC, gnoMAD, and 1000 genomes genetic variant databases.
The mutation is located in the Tubulin C-terminal domain (Figure 3). We tabulate a summary of phenotypic information from the 46 pathogenic and likely pathogenic variants with phenotypic information in TUBA1A tubulin C-terminal domain, which are present in clinvar, and 24 of the same variants and their more detailed phenotypes as they were reported by Tantry et al. (2023) [16]) and our case (Table 2).
3.3. Analysis of Mutation Location in the 3D Structure of Tubulin α-1A Chain Protein in a Complex with β-Tubulin
To explore our hypothesis of pathogenicity of the identified TUBA1A mutation, we sought to understand the possible effects of the His283Arg mutation on the structure of Tubulin α-1A Chain in a 3D structure of tubulin filaments. To do this, we performed protein visualization with PyMOL [17] on the published Cryo-EM structure of recombinant neuronal human tubulin 5JCO [18] to observe the Histidine 283 residue of interest and map its potential interactions. The location of the Histidine (residue 283) and the hydrogen bonds that it makes were marked on the α-Tubulin monomers in the 3D structure (Figure 4). Based on the published 3D Cryo-EM structure [18], the Histidine 283 residue is predicted to form two polar contacts: one as part of an innate backbone–backbone hydrogen bond with a neighbouring lysine at 280 (Lys280), and the second, an R-group, mediated polar contact through its imidazole ring with glutamine 85 (Gln85) of an adjacent α-tubulin subunit (Figure 4c). The second of these contacts would not be possible with α-tubulin that had an R-group substitution on residue 283 like the mutation of histidine to arginine found in our patient. This could cause a defect in lateral microtubule protofilament assembly, making it highly disruptive and, thus, pathogenic.
4. Discussion
TUBA1A encodes α-tubulin, a structural protein which plays a critical role in regulating neuronal migration in brain development [1]. Heterozygous de novo TUBA1A mutations cause an autosomal dominant “tubulinopathy”, with clinical features including brain malformations, microcephaly, neurodevelopmental delay, motor impairment, cognitive deficit, and epilepsy [1,21,22]. In this study, we identified a very rare TUBA1A mutation, His283Arg, located in the tubulin C-terminal domain (aa 263–392), in an infant that exhibited canonical tubulinopathy phenotypes with additional clinical features including some not previously reported with mutation of the gene.
TUBA1A is a highly constrained gene with very few missense mutations appearing in “healthy” population data. It has a Z-score of 8.75, indicating that it is very intolerant to amino acid changing variations without accompanying pathogenesis [23,24]. A total of 150 pathogenic and likely pathogenic variants with phenotypic information in TUBA1A are recorded in clinvar. Of these, almost a third (46) are in the tubulin C-terminal domain, between amino acids 263 and 392. In a recent review on the role of α tubulin isotypes in early brain development, Tantry et al. (2023) reported 77 published mutations, of which 24 were found in the same tubulin C-terminal domain [16]. The clinical features of mutations in this domain closely resemble those seen in our case. A comparison of phenotypic information for the tubulin C-terminal domain from our case, clinvar, and Tantry et al. (2023) is as shown in Table 2.
In consistency with tubulinopathy cases in clinvar and Tantry et al. (2023), our case displayed features of lissencephaly, numerous brain malformations, and seizures/epilepsy. Unique to our case were the phenotypes of Dandy Walker malformation/ventricular dilation, macrocephaly, hypotonia, and hypoplastic genitalia with a micropenis. Hebebrand et al. (2019) observed that prenatally diagnosed fetal cases are more severe than those diagnosed after birth, which is consistent with our case, which was diagnosed in utero.
As seen in Table 3, the majority of phenotypic features seen in our patient, except for macrocephaly, are also present in Walker–Walberg syndrome, which has the additional feature of retinal dysplasia. This overlap makes an accurate pre-term diagnosis very difficult without accompanying genetic analyses.
Using the published 3D structure of neuronal tubulin [18], we visualized the location of residue 283, Histidine, in the repeating α-tubulin 1A chain monomers (Figure 4a,b). Based on the published 3D structure, the imidazole ring of Histidine 283 is predicted to interact with Glutamine (residue 85) on the adjacent α-tubulin monomer in the filament through hydrogen bonding (Figure 4c). This hydrogen bond is predicted to be disrupted when the Histidine is mutated to Arginine (p.H283R) in the patient. Disrupting this hydrogen bonding, we predict, will impair the lateral interaction and, thereby, binding between adjacent α-tubulin monomers. This 3D modelling and the extreme rarity of the mutations are highly suggestive of a high level of pathogenicity.
Also, the novel clinical features seen in the infant are macrocephaly (rather than the more common microcephaly) and hypoplastic genitalia. The patient also exhibited a WWS-like generalized hypotonia, complex brain malformations, and seizures, which are also commonly seen in patients with TUBA1A mutations [2]. This infant was initially described as having clinical features resembling WWS, a severe form of autosomal recessive muscular dystrophy caused by mutations in glycosylation pathway genes. Clinical features of the syndrome include severe brain and eye abnormalities, as well as generalized severe hypotonia, muscle weakness, and seizures. Some of the clinical features overlap those associated with mutation of TUBA1A, highlighting the importance of determining a genetic diagnosis as early as possible (Table 3).
5. Conclusions
The TUBA1A mutation identified in the patient, c.848A>G His283Arg, is de novo, being absent in both parents. The mutation is ultra rare in clinical databases and not present in control genetic databases, which indicate that it is most likely deleterious. Analyses using in silico prediction tools and ACMG guidelines classified the mutation as pathogenic. The mutation is in the tubulin C-terminal domain, which also contains 46 other pathogenic and likely pathogenic variants that are associated with similar patient phenotypes. Based on 3D modelling of the histidine 283 residue, in the α tubulin structure, H283R is predicted to form an R-group-mediated polar contact through its imidazole ring with glutamine 85 (Gln85) of an adjacent a-tubulin subunit. The mutation of the histidine residue to arginine, in our case, is postulated to disrupt this and, thereby, disrupt the intermolecular interactions between adjacent α-tubulin-1A chain monomers in the 3D structure of microtubule filaments.
Together, the data support the TUBA1A mutation c.848A>G His283Arg as being causative of the neurological disorder and early death of the infant reported here. Genetic diagnosis of the patient allowed for genetic counselling to be offered to the family, including the risk of disease to other family members. Functional studies of the novel TUBA1A mutation, while challenging, may be warranted to determine the effects of the mutation on early brain development. Patients presenting with hypotonia, macrocephaly, and hypoplastic genitalia as well as brain malformations and seizures may not have WWS and should be considered for genetic testing for a possible tubulinopathy particularly involving the TUBA1A gene.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hebebrand M. Huffmeier U. Trollmann R. Hehr U. Uebe S. Ekici A.B. Kraus C. Krumbiegel M. Reis A. Thiel C.T. The mutational and phenotypic spectrum of tuba 1a-associated tubulinopathy Orphanet J. Rare Dis.2019143810.1186/s 13023-019-1020-x 30744660 PMC 6371496 · doi ↗ · pubmed ↗
- 2Buscaglia G. Northington K.R. Aiken J. Hoff K.J. Bates E.A. Bridging the gap: The importance of tuba 1a α-tubulin in forming midline commissures Front. Cell Dev. Biol.2021978943810.3389/fcell.2021.78943835127710 PMC 8807549 · doi ↗ · pubmed ↗
- 3Dobyns W.B. Pagon R.A. Armstrong D. Curry C.J. Greenberg F. Grix A. Holmes L.B. Laxova R. Michels V.V. Robinow M. Diagnostic criteria for walker-warburg syndrome Am. J. Med. Genet.19893219521010.1002/ajmg.13203202132494887 · doi ↗ · pubmed ↗
- 4Vajsar J. Schachter H. Walker-warburg syndrome Orphanet J. Rare Dis.200612910.1186/1750-1172-1-2916887026 PMC 1553431 · doi ↗ · pubmed ↗
- 5Beltran-Valero de Bernabe D. Currier S. Steinbrecher A. Celli J. van Beusekom E. van der Zwaag B. Kayserili H. Merlini L. Chitayat D. Dobyns W.B. Mutations in the o-mannosyltransferase gene pomt 1 give rise to the severe neuronal migration disorder walker-warburg syndrome Am. J. Hum. Genet.2002711033104310.1086/34297512369018 PMC 419999 · doi ↗ · pubmed ↗
- 6Beltran-Valero de Bernabe D. Voit T. Longman C. Steinbrecher A. Straub V. Yuva Y. Herrmann R. Sperner J. Korenke C. Diesen C. Mutations in the fkrp gene can cause muscle-eye-brain disease and walker-warburg syndrome J. Med. Genet.200441 e 6110.1136/jmg.2003.01387015121789 PMC 1735772 · doi ↗ · pubmed ↗
- 7Riemersma M. Mandel H. van Beusekom E. Gazzoli I. Roscioli T. Eran A. Gershoni-Baruch R. Gershoni M. Pietrokovski S. Vissers L.E. Absence of α- and β-dystroglycan is associated with walker-warburg syndrome Neurology 2015842177218210.1212/WNL.000000000000161525934851 · doi ↗ · pubmed ↗
- 8Hercus C. Novoalign v 4.4Novocraft Technologies. Linux 2022 Available online: https://www.novocraft.com/support/download/(accessed on 27 July 2024)