Cytogenetically Balanced Reciprocal Translocation Could Hide Molecular Genomic Unbalances: Implications for Foetal Phenotype Correlation
Nicoletta Villa, Serena Redaelli, Stefania Farina, Elena Sala, Francesca Crosti, Sabrina Cozzolino, Maria Verderio, Leda Dalprà, Gaia Roversi, Angela Bentivegna, Giovanni Cazzaniga, Marialuisa Lavitrano, Donatella Conconi

TL;DR
A seemingly balanced genetic change in a fetus can actually hide a genomic imbalance, affecting prenatal diagnosis and risk assessment.
Contribution
Highlights the need for molecular testing alongside karyotype analysis in prenatal diagnosis of chromosomal abnormalities.
Findings
A cytogenetically balanced translocation was found to be genomically unbalanced.
The case shifted the risk from Down syndrome to Zellweger syndromic spectrum due to a PEX3 deletion.
A healthy baby was born despite the initial concerns.
Abstract
When an increased nuchal translucency (>3.00 mm) is observed during the echographic examination of a foetus in the first trimester of pregnancy, an increased risk of chromosomopathy is considered, and the pregnant woman is offered the possibility of an invasive investigation. Here, we focused our attention on prenatal diagnosis issues in cases of foetuses with cytogenetically balanced reciprocal translocations. We report the finding of a cytogenetically balanced, de facto genomically unbalanced translocation that poses a challenge in a case of prenatal diagnosis, changing the risk of Down syndrome in a Zellweger syndromic spectrum risk (PEX3 deletion). At term, a healthy baby was born. This case teaches that prenatal diagnosis in cases of foetuses at increased risk of chromosomal abnormality imperatively requires molecular investigation in addition to a morphological karyotype.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
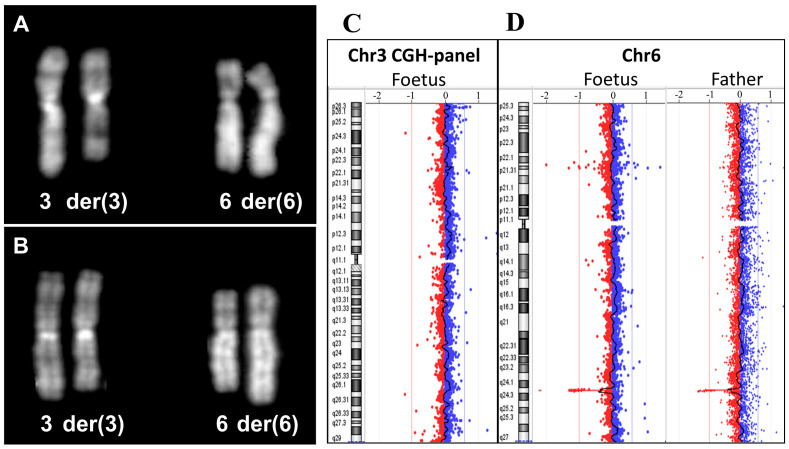 Figure 1
Figure 1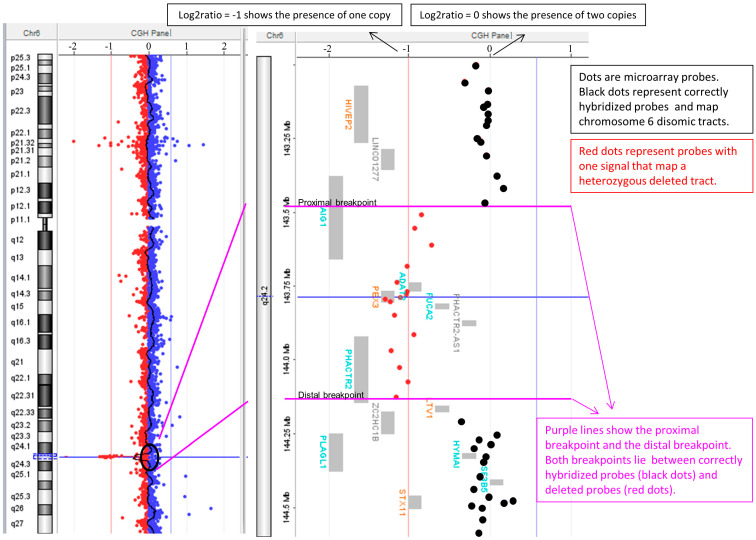 Figure 2
Figure 2- —the University of Milano-Bicocca
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPrenatal Screening and Diagnostics · Genomic variations and chromosomal abnormalities · Cancer Genomics and Diagnostics
When an increased nuchal translucency (>3.00 mm) is found in a foetus during the first trimester, even as the only clinical indication (isolated indication, alarming for the health condition of the foetus itself), the pregnant woman is offered the possibility of invasive investigation such as chorionic villus sampling to verify the foetal karyotype.
In the literature for these cases, numerous articles report the frequency of karyotype abnormalities, which is extremely variable between 0.5% and 6.6% [1]. Moreover, it is emphasised that the use of a comparative genomic hybridisation (CGH) array in foetuses with a normal karyotype detects on average an extra 5% of pathological copy number variations (CNVs). Normally, in these cases, a trisomy is expected, with trisomy 21 as the most frequent, but it is possible that structural abnormalities of the chromosomes, such as translocations or rare genetic/genomic conditions, are evidenced. Numerous genetic disorders have in fact been identified in association with the increase in nuchal translucency [2,3,4]; these include, for example, 22q11 micro-deletion syndrome, Noonan syndrome, and Smith–Lemli–Opitz syndrome [1].
We report the finding of a cytogenetically balanced, de facto genomically unbalanced translocation that poses a challenge in a case of prenatal diagnosis, changing the risk for Down syndrome in a Zellweger syndromic spectrum risk (Figure 1 and Figure 2).
CGH array analysis was also performed on both parents. The mother had a normal molecular karyotype, while the father showed a deletion in the same band of the cytogenetic translocation breakpoint (D). In all the consulted databases, no similar deletions were reported, so the CNV was considered as clinically uncertain (VOUS) because the foetal morphology was apparently normal and also because it was present in the molecular karyotype of the phenotypically normal father. The final foetal karyotype was as follows:
46,XY,t(3;6)(q25.3;q25.1)pat.arr[GRCh37] 6q24.2(143512294_144136217)x1 pat.
The lost genes in the deletion were the following: AIG1 (exons from 4 to 6), ADAT2, PEX3 (OMIM613164), FUCA2, PHACTR2-AS1, and PHACTR2 (exons from 1 to 12).
To verify if the chromosome 6 alleles were biparentally inherited, thus excluding the uniparental disomy of chromosome 6 that is associated with transient neonatal diabetes mellitus [5,6], six microsatellites mapped in 6q13-q26 were tested. The analysis showed evidence of biparental allelic heredity.
The pregnancy progressed normally, and the foetus underwent obstetric ultrasound scans every 4-6 weeks that consistently documented regular foetal anatomy and foetal biometry at the upper limits, typical in the third trimester. The baby was born at 38+2 gestational weeks via caesarean section for breech presentation. The parameters at birth were as follows: Apgar at 1′ and 5′ minute: 8 and 9; weight: 3330 g; length: 49 cm (32nd percentile); CC: 35.5 cm (80th percentile). Nothing was reported at the first paediatric visit. After 18 months of life, the baby was in perfect health and the mother reported that they are very bright.
It is known that an increased thickness of the foetal nuchal fold is an “indicator” of risk for chromosomal/genetic abnormality [1,3]. Hence, following counselling, the mother was given the option to perform a placenta biopsy and both morphological and molecular karyotyping. This is because it has been established that the CGH array adds approximately 5% of pathological variants in the presence of a normal chromosomal karyotype [1]. In the case presented here, the biochemical test indicated both a particularly increased risk of numerical chromosomal abnormalities and an increase in nuchal translucency. The result of the genetic investigations showed a translocation between chromosome 3 and chromosome 6 of paternal origin, associated with a molecular deletion containing the PEX3 gene.
De Gregori et al. described cryptic genomic abnormalities in 59 patients showing clinical phenotypes and a balanced karyotype [7]. Moreover, phenotypically normal individuals showing an apparently balanced translocation, at deep analysis with CGH array, were found to have an imbalance with gene disruption [8].
PEX3 (OMIM613164) gene maps in 6q24.2 and the genomic coordinates are from nt 143,771,942 to nt 143,811,753 (GRCh37 assembly). In 6q24.2 micro-deletion, the PEX3 gene is completely included, so one copy of the gene is lost. At the extremities of the deletion, the two interrupted genes (AIG1 and PHACTR2) are not associated with pathology and are not dose-sensitive. The mutations in the PEX3 gene are associated with disease risk in an autosomal recessive manner and PEX3 is a moderately dose-sensitive gene.
In Decipher Database, the PEX3 haploinsufficiency score (HI) is 27.6% (values from 0 to 10% mean “more likely haploinsufficient gene”, from 90 to 100% “more likely to not exhibit haploinsufficiency”), while the probability of Loss-of-function (LOF) Intolerance (pLI) is 0.02 (values ≥ 0.9 LOF not tolerated, ≤0.1 LOF tolerated). These values collectively suggest that this gene does not appear to be haploinsufficient and that heterozygous mutations do not seem to have deleterious effects.
Peroxins are essential for peroxisome function, and genetic abnormalities that alter their biogenesis constitute an autosomal recessive heterogeneous group of diseases known as Zellweger spectrum disorders (ZSD), characterised by the absence or reduction in functional peroxisomes in cells. Due to the phenotypic variability, ZSD was originally described as multiple distinct syndromes including Zellweger syndrome (ZS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), rhizomelic chondrodysplasia punctata type 1 (RCDP1), and Heimler syndrome [9,10,11]. Based on a common peroxisomal basis, these disorders are now identified as ZDS, ranging from severe, intermediate, and milder phenotypes [10].
Disease severity often coincides with the age at which symptoms first appear [12]. Neonatal presentation is associated with a severe phenotype, with hypotonia, reduced spontaneous movements, feeding problems, seizures, direct hyperbilirubinemia, and elevated liver enzymes. Facial dysmorphism, large fontanelles, wide sutures, hypoplastic supraorbital ridges, and a broad nasal bridge are often reported. Ocular abnormalities include glaucoma, cataracts, and retinopathy. Death generally occurs within the first year of life [9,13].
Childhood presentation is associated with developmental delay, failure to thrive, retinal dystrophy, sensorineural hearing loss, feeding problems, hepatic dysfunction, and adrenal insufficiency. A regression of previously attained neurological milestones can occur secondary to demyelination (progressive leukodystrophy). Death may occur prior to adolescence [9,13].
In the adolescent and adult presentation of mild or absent developmental delay, neuroregression, cerebellar ataxia, peripheral neuropathy, adrenal insufficiency, and leukodystrophy can be observed. Life expectancy is variable [9,13].
The clinical spectrum of ZSD is due to mutations in one of the 13 PEX genes encoding peroxins. Pathogenic mutations in PEX1 are responsible for about 60% of ZS cases, while the literature shows that homozygous pathogenic variants in the PEX3 gene are responsible for only 0.7%; however, there are no data on deletions and/or duplications of the gene [14].
Since PEX3 is involved in less than 1% of cases, and considering an incidence of ZS of 1/133000 (NY State newborn screening), the risk that the described foetus may be affected by ZS depends on the probability that the mother is a healthy carrier of a mutation in the same gene. The calculation of the healthy carrier in the general population for PEX3 mutation is about 1 in 1820; therefore, the theoretical risk of ZS for the foetus, which is already a carrier of the paternal PEX3 deletion, is approximately 1 in 3640 (0.03%).
Generally, prenatal diagnosis for ZSD is feasible if parental variants are known; a carrier test for PEX3 mutations is technically feasible, but gene sequencing is burdened with too many variants of unknown significance, and is not clinically actionable, especially in prenatal context. Otherwise, biochemical screening assays in body fluids, i.e., amniotic, could be performed, but with limited diagnostic sensitivity. Since the “a priori” risk of the affected foetus was very low, the foetal development was normal with normal morphology at ultrasound examination, and, not in the least due to the high risk for chromosomal unbalanced conceptions due to an advanced maternal age and paternal translocation, the parents decided to continue the pregnancy. No foetal anomalies were evidenced at accurate ultrasound follow up and a healthy baby was born at term. No genetic tests were suggested for the reproductive risk related to Zellweger syndrome because the mother was a known carrier of the BRCA2 mutation and underwent ovariectomy.
In conclusion, this case teaches that prenatal diagnosis in cases of foetuses at an increased risk of chromosomal abnormality imperatively requires molecular investigation, as does the morphological karyotype. Both complement each other, with one showing structural abnormalities and the other highlighting the loss and/or duplication of regions or individual gene anomalies below the level of resolution of classical cytogenetics.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Grande M. Jansen F.A.R. Blumenfeld Y.J. Fisher A. Odibo A.O. Haak M.C. Borrell A. Genomic microarray in fetuses with increased nuchal translucency and normal karyotype: A systematic review and meta-analysis Ultrasound Obstet. Gynecol.20154665065810.1002/uog.1488025900824 · doi ↗ · pubmed ↗
- 2Petersen O.B. Smith E. Van Opstal D. Polak M. Knapen M.F.C.M. Diderich K.E.M. Bilardo C.M. Arends L.R. Vogel I. Srebniak M.I. Nuchal translucency of 3.0-3.4 mm an indication for NIPT or microarray? Cohort analysis and literature review Acta Obstet. Gynecol. Scand.20209976577410.1111/aogs.1387732306377 PMC 7318216 · doi ↗ · pubmed ↗
- 3de Wit M.C. Srebniak M.I. Govaerts L.C. Van Opstal D. Galjaard R.J. Go A.T. Additional value of prenatal genomic array testing in fetuses with isolated structural ultrasound abnormalities and a normal karyotype: A systematic review of the literature Ultrasound Obstet. Gynecol.20144313914610.1002/uog.1257523897843 · doi ↗ · pubmed ↗
- 4Souka A.P. Von Kaisenberg C.S. Hyett J.A. Sonek J.D. Nicolaides K.H. Increased nuchal translucency with normal karyotype Am. J. Obstet. Gynecol.200519210051021 Erratum in Am. J. Obstet. Gynecol. 2005, 192, 209610.1016/j.ajog.2004.12.09315846173 · doi ↗ · pubmed ↗
- 5Temple I.K. Mackay D.J.G. Diabetes Mellitus, 6q 24-Related Transient Neonatal Gene Reviews® Adam M.P. Feldman J. Mirzaa G.M. Pagon R.A. Wallace S.E. Bean L.J.H. Gripp K.W. Amemiya A. University of Washington Seattle, WA, USA 200520301706 · pubmed ↗
- 6Greeley S.A.W. Polak M. Njølstad P.R. Barbetti F. Williams R. Castano L. Raile K. Chi D.V. Habeb A. Hattersley A.T. ISPAD Clinical Practice Consensus Guidelines 2022: The diagnosis and management of monogenic diabetes in children and adolescents Pediatr. Diabetes 2022231188121110.1111/pedi.1342636537518 PMC 10107883 · doi ↗ · pubmed ↗
- 7De Gregori M. Ciccone R. Magini P. Pramparo T. Gimelli S. Messa J. Novara F. Vetro A. Rossi E. Maraschio P. Cryptic deletions are a common finding in “balanced” reciprocal and complex chromosome rearrangements: A study of 59 patients J. Med. Genet.20074475076210.1136/jmg.2007.05278717766364 PMC 2652810 · doi ↗ · pubmed ↗
- 8Baptista J. Prigmore E. Gribble S.M. Jacobs P.A. Carter N.P. Crolla J.A. Molecular cytogenetic analyses of breakpoints in apparently balanced reciprocal translocations carried by phenotypically normal individuals Eur. J. Hum. Genet.2005131205121210.1038/sj.ejhg.520148816118644 · doi ↗ · pubmed ↗