No evidence that ACE2 or TMPRSS2 drive population disparity in COVID risks
Nathaniel M. Pearson, John Novembre

TL;DR
This paper challenges the idea that genetic differences in ACE2 and TMPRSS2 genes explain population disparities in COVID risks, showing that earlier claims were based on flawed data interpretation.
Contribution
The paper identifies a sampling artifact in prior research and provides empirical evidence that other genetic loci are more significant for COVID risk than ACE2 and TMPRSS2.
Findings
Earlier claims about ACE2 and TMPRSS2 variants driving population disparities in COVID risk were based on flawed data interpretation.
Empirical evidence shows that other genetic loci have a more significant impact on personal COVID risks than ACE2 and TMPRSS2.
Variation in ACE2 and TMPRSS2 is unlikely to exacerbate population disparities in the effects of more risk-informative loci.
Abstract
Early in the SARS-CoV2 pandemic, in this journal, Hou et al. (BMC Med 18:216, 2020) interpreted public genotype data, run through functional prediction tools, as suggesting that members of particular human populations carry potentially COVID-risk-increasing variants in genes ACE2 and TMPRSS2 far more often than do members of other populations. Beyond resting on predictions rather than clinical outcomes, and focusing on variants too rare to typify population members even jointly, their claim mistook a well known artifact (that large samples reveal more of a population’s variants than do small samples) as if showing real and congruent population differences for the two genes, rather than lopsided population sampling in their shared source data. We explain that artifact, and contrast it with empirical findings, now ample, that other loci shape personal COVID risks far more significantly…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
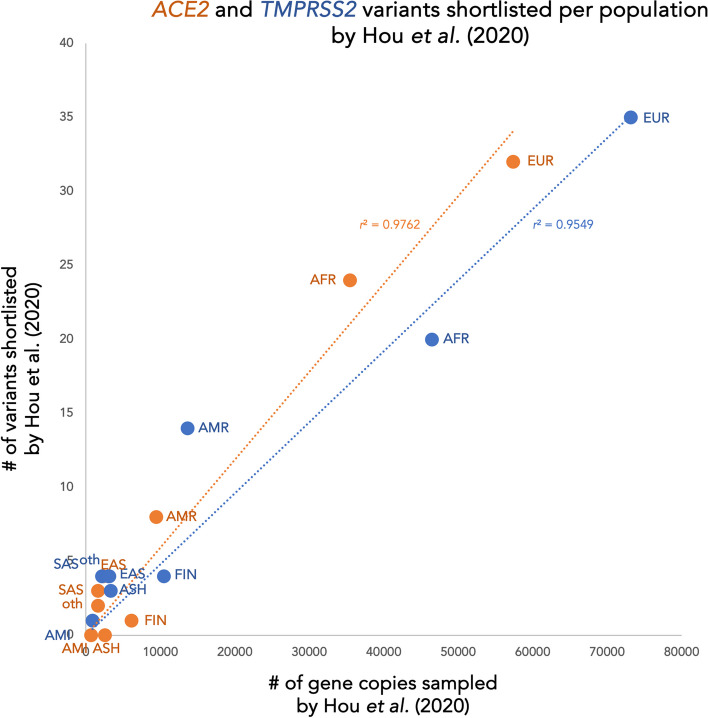 Figure 1
Figure 1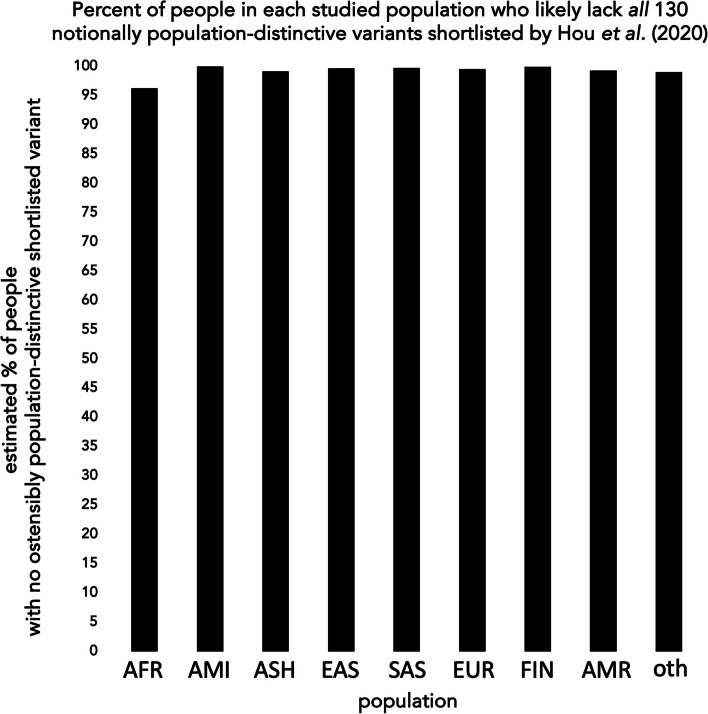 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSARS-CoV-2 and COVID-19 Research · COVID-19 Clinical Research Studies · Genetic Associations and Epidemiology
Background
In mid-2020, concurrent with early empirical inquiry into roles of host genomic variation in SARS-CoV2 infection [1–5], Hou et al. set out to offer predictive guidance for such efforts, by assessing pre-pandemic public DNA data from two human genes, ACE2 and TMPRSS2, whose protein products were known to interact with other coronaviruses [6].
Pooling public genotype data sampled from various human groups, without phenotypes, they shortlisted ACE2 and TMPRSS2 variants that some computational heuristics predicted likely to alter protein function, and found that most such variants (each, typically, very rare) came from subsets of data labeled “African/African-American” or “Non-Finnish European” versus labeled otherwise (e.g., “East Asian”).1 This, they held, suggested real-world population disparities in ACE2 and TMPRSS2 functional variant load, similar for both genes, that might in turn drive population differences in COVID outcomes.
Errant interpretation of genotype data
Alas, Hou et al. had neglected a basic feature of the public data they used—lopsided population sample sizes—that made their summary findings artifactually likely even with no difference between real populations. Specifically, they had pooled genotypes from > 36,000 “non-Finnish European” and > 23,000 “African/African-American” people, but far fewer “Amish” (450), “Ashkenazi” (1662), “East Asian” (1567), or other (< 15,000) people.2 As such, even if variants were uniformly distributed across real populations, Hou et al. would likelier find a given rare variant in either of their big samples (“African/African-American” or “non-Finnish European”) than in any of their much smaller samples of other groups.
Consistent with such artifact, the number of ACE2 or TMPRSS2 variants Hou et al. shortlisted for a given population scales well with how many genotypes they sampled from that population (Fig. 1, origin-rooted linear r^2^ > 0.95 for both genes).3^,^4 Long known in theoretical and empirical population genetics [7, 11], the sampling effect apparent in Hou et al.’s summary findings reflects a simple fact: much as counting more of a forest’s birds can help document rare taxa living there, sequencing more of a population’s gene copies helps document rare variants among them. While other factors shape the emergence and fate of such variants, and their rate of discovery with increasing sample size [7–10, 12, 13], reliably finding and quantifying them entails sampling from many individuals.Fig. 1. Population-specific variant tallies in Hou et al. [6] reflect lopsided sampling. Scatterplots of population-specific tallies (y-axis) of shortlisted variants in ACE2 (orange) or TMPRSS2 (blue), by sample size (x-axis; values denote maximum sampled alleles among shortlisted variant-position genotypes for that gene in gnomAD (v.3.0) + NHLBI-GO ESP6500 genotypes, as pooled by Hou et al. [6]). Datapoints mark values for African/African-American (AFR; TMPRSS2 count excludes 1 variant (p.Pro444Leu) reported by Hou et al., but not in public data, and not consistent with reference variant at given protein residue); Amish (AMI); Ashkenazi (ASH); east Asian (EAS); south Asian (SAS); non-Finnish, non-Amish, non-Ashkenazi European (EUR; TMPRSS2 count excludes 1 variant (p.Gly6Arg) reported by Hou et al., but not in public data, and not consistent with reference variant at given protein residue); Finnish (FIN in Hou et al. [6]); Latino/Admixed American (AMR; ACE2 count includes 2 variants wrongly omitted from this population by Hou et al.); or other (oth; PNA in Hou et al. [6]; ACE2 count excludes 2 variants wrongly tallied in this population by Hou et al.). Best-fit trends (dashed) mark origin-rooted linear regression, conservatively proxying independent (versus cumulative) discovery of potentially selection-constrained (versus selectively neutral) variants in samples from variably sized, mutually diverged populations (versus one steady-sized randomly mating population). We note that even in the contrasting case of cumulative discovery in a steady-sized population, variants under selective constraint (as Hou et al. sought to tally) tend to accrue quasi-linearly, rather than strictly logarithmically, with increasing overall sample size [8–11]
As such, current public genotype data inherently document more of the variants actually circulating in some populations than of those actually circulating in other populations—and allow more precise estimation, in the same best-sampled (if at all) groups, of each variant’s actual frequency (as may figure in functional prediction) or absence. For example, to be 90% confident that even the least rare shortlisted ACE2 variant in non-Finnish, non-Amish, non-Ashkenazi European sample data (p.R219C) is not actually more common in Amish, Ashkenazi, and/or Finnish populations, despite its absence in those groups’ small sample data, would require sampling > 22,700 additional Amish, Ashkenazi, and Finnish copies of ACE2.5
Beyond summary tallies, none of Hou et al.’s shortlisted variants reliably proxies any population to begin with. One (TMPRSS2 p.V160M) appears in all studied populations—and in many individuals in each—so offers scant ground to guess which population(s) a carrier comes from. All 130 other shortlisted variants appear too rare in every population to typify members of any of them (even in aggregate, their data suggest that > 96% of people in every studied population likely carry none of those rare variants (Fig. 2)).6 And as real populations also harbor unsampled but functionally relevant variants, whose effects on basic protein function (let alone response to a particular virus) current heuristics cannot reliably predict [14–16], the tallies and predictions of Hou et al. do not warrant positing that ACE2 or TMPRSS2 (let alone both) functions worse with respect to SARS-CoV2 in any human population (let alone particular ones) than in others.Fig. 2. Nearly everyone, in all studied populations, likely lacks all ostensibly population-distinctive variants shortlisted by Hou et al. [6]. Bar plot of estimated percentage of people in each studied population who likely have none of the 130 notionally population-distinctive (i.e., absent in sample data from at least one studied population) ACE2 and TMPRSS2 variants shortlisted (without empirical evidence for any effect on protein function or other physiology, and omitting many other potentially functionally relevant variants in all populations) by Hou et al. [6]. Estimates (product of binomial probabilities) presume variants assort randomly, independently, at sampled population-specific frequencies, in half-XX/half-XY populaces. AFR = African/African-American; AMI = Amish; ASH = Ashkenazi; EAS = east Asian; SAS = south Asian; EUR = non-Finnish, non-Amish, non-Ashkenazi European; FIN = Finnish; AMR = Latino/Admixed American; oth = other. Values may underestimate true minimum region-wide percentage, as (i) the least rare such variant (ACE2 p.L731F), which most strongly suppresses the AFR estimate, appears mainly in data from over-proportionately sampled west Africa, more so than in data yet sampled from likewise populous peoples elsewhere in Africa and diaspora [17]; and (ii) any pairwise linkage among shortlisted variants would increase the proportion of people inheriting neither variant in such pairs
In principle, real populations may indeed differ, if modestly, in functionally relevant patterns of variation in a gene (or even genome-wide), pending their histories. All else equal, for example, big populations tend to accrue and keep more genetic variation (especially if non-harmful) than do small populations [12, 18, 19]; fast-growing populations to accrue more new rare variation in particular [9, 20]; and genome segments under strong directional selection, in a given population’s environment, may (along with flanking segments coupled to them without recombination) tend to lose such rare variation in particular [10].
But as human genes thus vary in summary patterns of variation, via potentially population-distinctive histories, meaningfully comparing such patterns requires sampling well, and assessing not just which variants appear at all in data sampled from a population, but the summary distributions of their estimated frequencies (respective proportions of sampled gene copies harboring them).
To that end, frequency-sensitive summary metrics7 show less variation in human ACE2, both within and between most human populations,8 than for most other X-borne9 or autosomal [21, 22, 28, 29] human genes, limiting the extent to which populations’ distinctive histories may yield disparate patterns of variation. By comparison, such well grounded summary metrics show more overall variation in human TMPRSS2 [21]—much of it shared across populations, in varied patterns that reflect the cross-regional spread of variants old (and generally non-harmful) enough to have become common.
Importantly, even beyond the two genes’ contrasting patterns of variation, pandemic-long cohort outcomes have not shown variation in either ACE2 or TMPRSS2 to shape personal COVID risks nearly as significantly as variation elsewhere in our genomes—including the most strongly and significantly risk-shaping locus, on the short arm of chromosome 3; the ABO blood group locus on chromosome 9; and other autosomal loci [1–5, 17, 30]. Some non-protein-altering variants in ACE2 and TMPRSS2 have met multiple-test-stringent significance criteria for association with risks of SARS-CoV2 infection (an ACE2 regulatory variant cluster) or severity (TMPRSS2 intronic variant), but their significance falls short of that evident for other loci. And among variants shortlisted by Hou et al., only one (the relatively common TMPRSS2 p.V160M) has shown even suggestive (not multiple-test-stringent) evidence for association with any COVID risk [30–33]—while broader tests, tuned and powered specifically to detect rare variant association per se in clinically characterized population cohorts, have not implicated shortlisted or other rare protein-coding variation in either gene in COVID risks [34, 35].
Moreover, across all COVID-implicated loci, risk-informative variants differ in their population distributions and inferred effects, in many cases in partial counter-balance to one another. The ACE2-regulatory rs190509934C variant, for example, associates significantly with below-average risk of SARS-CoV2 infection (and suggestively with below-average risk of severe COVID), but appears least rare in a studied broad population (south Asian) in which other loci harbor variant loads most strongly associated with above-average risk of infection (and severe disease) [36, 37].
Altogether, such mixes of above- and below-average risk-associated variants in human genomes worldwide leave a broad range of risk-relevant personal variant load within every studied human population [38–41]. And those loads, in turn, explain < 10% of personal variability in COVID risks evident, so far, in clinically characterized cohorts [39], while other factors, such as age, background health, and immune exposures, show far stronger effects.10 As such, now-ample data suggest that COVID incidence and severity likely differ among human populations far less by genetics than by factors such as age structure, past and ongoing immune exposures, comorbidity prevalences, and access to effective health interventions [4, 17, 30, 39, 40, 45–52]—and pointedly, do not support speculation that variation in ACE2 or TMPRSS2 drives net population disparity in genetically attributable (or overall) COVID risks.11
Conclusions
Speculating early in the pandemic, on potential COVID-relevance of variation in two of humanity’s many genes, Hou et al. understandably settled for predictive heuristics in lieu of clinical data. But in tallying shortlisted ACE2 and TMPRSS2 variants among populations, they mistook a sample size artifact as if evidence of population differences—and tried to proxy population-representative gene function by tallying shortlisted variants found at all, instead of summing empirically estimated, genotype frequency-scaled effects. Though their shortlist offered well intended, if unvalidated, candidates for early-pandemic study, genome-wide empirical insights have eclipsed its utility—while leaving, unaddressed, their artifactual summary claims. And those claims, in turn, have drawn credulous citation in public discourse, hindering understanding of COVID risks and of human genetic diversity itself [57–59].
While pitfalls of methodology and interpretation have long plagued basic and clinical research [60–67], public discourse invoking Hou et al. [6] highlights how platforms to usefully share and discuss such research can also virally spread faulty inferences missed by authors and reviewers, misleading not just researchers and clinicians, but also lay-people who may rely on published science in personal, professional, family, and civic decisions [64]. As such, we hope that correcting basic errors that misled Hou et al., and those citing their work, helps right the record on human genomic variation in ACE2, TMPRSS2, and loci more informative of COVID risks—and, further, encourages critical stringency in the interpretation of population genetic and other data, amid efforts [68, 69] to more promptly and soundly validate research published, and subsequently invoked, in and between societal crises.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Gunning AC, Fryer V, Fasham J, Crosby AH, Ellard S, Baple E, et al. Assessing performance of pathogenicity predictors using clinically-relevant variant datasets. bio Rxiv. 2020;:2020.02.06.937169.10.1136/jmedgenet-2020-107003 PMC 832732332843488 · doi ↗ · pubmed ↗
- 2Zhang C, Verma A, Feng Y, Dos Reis Melo MC, Mc Quillan M, Hansen M, et al. Impact of natural selection on global patterns of genetic variation, and association with clinical phenotypes, at genes involved in SARS-Co V-2 infection. Res Sq. 2021. 10.21203/rs.3.rs-673011/v 1.10.1073/pnas.2123000119 PMC 917376935580180 · doi ↗ · pubmed ↗
- 3Covid-19 HGI Browser. https://app.covid 19hg.org/variants. Accessed 4 Aug 2023.
- 4gnom AD. https://gnomad.broadinstitute.org/. Accessed 23 Oct 2023.
- 5Hodgman L, Kennedy RF Jr. denies comments on ‘ethnically targeted’ COVID-19 were anti-Semitic. POLITICO. https://www.politico.com/news/2023/07/15/rfk-jr-covid-19-ethnically-targeted-00106478. Accessed 6 Aug 2023.
- 6Kennedy RF Jr. [...]a 2021 study of the COVID-19 virus [sic] shows that COVID-19 appears to disproportionately affect certain races […] That study is here: https://pubmed.ncbi.nlm.nih.gov/32664879/ Twitter. 2023. https://twitter.com/Robert Kennedy Jr/status/1680227322509635595. Accessed 4 Aug 2023.
- 7Owens C, Kennedy RF Jr. mentioned an actual scientific study which revealed that Chinese people & Ashkenazi Jews were the most immune to COVID-19 [...]. https://bmcmedicine.biomedcentral.com/articles/10.1186/s 12916-020-01673-z … Twitter. 2023. https://twitter.com/Real Candace O/status/1680371443446722561. Accessed 6 Aug 2023.