Diagnostic Challenges in the Myopathic Variant of Carnitine Palmitoyltransferase II Deficiency: A Case Report
Lana Alabbasi, Hadhami Ben Turkia, Maram Nass, Ibrahim Sahin

TL;DR
A 10-year-old boy with a rare metabolic disorder showed exercise-induced muscle damage, and genetic testing identified a new mutation linked to the condition.
Contribution
The paper reports a novel compound heterozygous pathogenic variant in CPT2 associated with myopathic carnitine palmitoyltransferase II deficiency.
Findings
The patient's condition improved with hydration, glucose, carnitine, and alkalinization.
Genetic testing identified a compound heterozygous pathogenic variant c.338C>T and a variant of unknown significance c.729_731del.
The patient experienced three further attacks over four years despite dietary and lifestyle interventions.
Abstract
Carnitine palmitoyltransferase II deficiency is a rare metabolic disorder affecting the mitochondrial oxidation of fatty acids. We present a case of the myopathic form in a 10-year-old Bahraini male following an initial presentation of exercise-induced rhabdomyolysis and transaminitis. There was no consanguinity or findings suggestive of an underlying inborn metabolic disorder. Tandem mass spectrometry on dried blood spots showed no abnormal acyl-carnitines profile. The condition improved with hyperhydration, high glucose intake, carnitine, and alkalinization. Genetic testing revealed a compound heterozygous pathogenic variant c.338C>T (p.Ser113Leu) and a variant of unknown significance c.729_731del (p.Leu244del). The patient was kept on a high carbohydrate and low-fat diet with medium chain triglycerides supplementation and advised to avoid long fasting periods and strenuous exercise.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
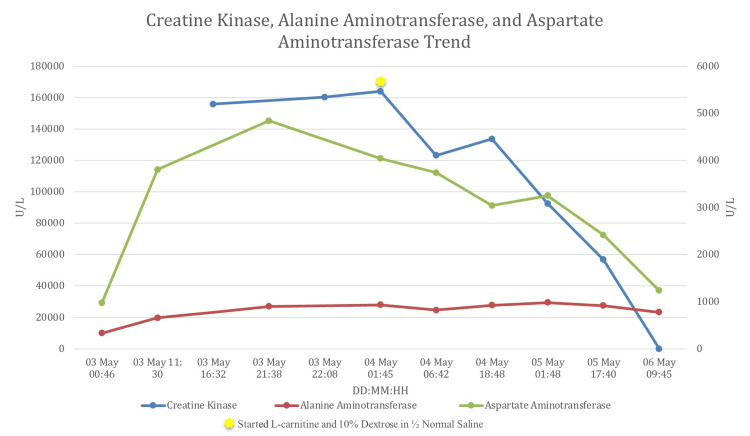 Figure 1
Figure 1| Test | Result | Reference value | |
| Bloods | White blood count | 11.42 × 109 /L | 4 - 11× 109/L |
| Hemoglobin | 13.8 g/dl | 11 - 15.5 g/dl | |
| Platelets | 275 × 109/L | 150 - 450 × 109/L | |
| C-reactive protein | 1 mg/l | 0 - 10 mg/l | |
| Random glucose | 6.9 mmol/l | 3.6 - 8.9 mmol/l | |
| Sodium | 138 mmol/l | 137 - 148 mmol/l | |
| Potassium | 3.6 mmol/l | 3.5 - 5.1 mmol/l | |
| Urea | 6.9 mmol/l | 3 - 7 mmol/l | |
| Creatinine | 60.5 umol/l | 27 - 62 umol/l | |
| Total protein | 72.3 g/l | 64 - 82 g/l | |
| Albumin | 43.4 g/l | 30 - 50 g/l | |
| Globulin | 28.9 g/l | 23 - 35 g/l | |
| Bilirubin total | 8.4 umol/l | 1.71 - 20.5 umol/I | |
| Bilirubin direct | 2.09 umol/l | 0 - 5 umol/l | |
| Aspartate aminotransferase | 972.1 u/l | 10 - 40 u/l | |
| Alanine aminotransferase | 331 u/l | 16 - 63 u/l | |
| Alkaline phosphatase | 241 u/l | 10 - 14 years: 130 - 340 u/l | |
| Gamma-glutamyl transferase | 20.7 u/l | 0 - 35 u/l | |
| Hepatitis profile (A, B, and C) | Negative | Not applicable | |
| Activated partial thromboplastin ratio | 0.94 | 0.9 - 1.4 | |
| International normalized ratio | 0.94 | 0.61 - 1.17 | |
| Prothrombin time | 11.4 seconds | 10.7 - 13.9 seconds | |
| Lactic acid | 1.8 | < 2.15 | |
| Calcium | 2.1 mmol/l | 2.12 - 2.62 mmol/l | |
| Magnesium | 0.9 mmol/l | 07 - 1 mmol/l | |
| Inorganic phosphorus | 1.6 mmol/l | 0.80 - 1.40 mmol/L | |
| Creatine kinase | 155800 U/L | 35 - 232 U/L | |
| Creatine kinase myocardial band (MB) | 233.8 ng/ml | 0 - 5 ng/ml | |
| Troponin-1 | 0.03 ng/ml | 0.02 - 0.06 ng/ml | |
| Tandem mass spectrometry for amino acids, organic acids, and acylcarnitine | Normal | Not applicable | |
| Urine | Urine culture and sensitivity | Negative | Not applicable |
| Urine myoglobin | 242 ng/ml | < 20 ng/ml |
| Inborn errors of metabolism causing rhabdomyolysis | |
| Glycogen storage disorders | Glycogen storage disease type V |
| Disorders of glycolysis | Aldolase A deficiency |
| Lactate dehydrogenase deficiency | |
| Muscle phosphofructokinase deficiency | |
| Phosphoglycerate kinase deficiency | |
| Disorders of mitochondrial fatty acids oxidation | Carnitine palmitoyltransferase 2 deficiency (CPT2) |
| Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHAD) | |
| Very long-chain acyl-CoA dehydrogenase deficiency (VLCAD) | |
| Trifunctional protein deficiency (TF) | |
| Mitochondrial disorders oxidative phosphorylation system deficiencies | |
| Others | Lipin-1 deficiency (LPIN1) |
| Transport and Golgi organization 2 (TANGO2) | |
| Ryanodine receptor 1 (RYR1) | |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMetabolism and Genetic Disorders · Muscle and Compartmental Disorders · Neurological and metabolic disorders
Introduction
Mitochondrial beta-oxidation of fatty acids is one of the significant sources of energy, providing up to 80% of the total requirement during fasting or prolonged exercise. In such conditions, long-chain fatty acids (LCFAs), hexadecanoyl-L-carnitine (C16) to octadecenoyl-L-carnitine (C18) stored as triglycerides in fat tissue, are transported into cells and activated to acyl-CoA esters. The carnitine palmitoyltransferase II (CPT II) enzyme is an inner mitochondrial membrane protein that cleaves fatty acids from carnitine to be used in beta-oxidation resulting in acetyl-CoA production, which enters the Krebs cycle to produce energy or is converted to ketone bodies that can be used as alternative energetic fuel [1,2].
CPT II deficiency (CPT II-D) is an autosomal recessive disorder that can present with one of three phenotypes: lethal neonatal, infantile hepatocardiomuscular, or myopathic form. The former two types are severe multisystemic diseases characterized by hypoketotic hypoglycemia, cardiomyopathy, seizures, and early death. Around 20 families with the lethal neonatal form, 28 families with the severe infantile hepatocardiomuscular form, and 300 cases of the myopathic form have been reported. However, prevalence might be underestimated as many pregnancies with the lethal neonatal form end due to severe cerebral malformations, and symptoms of the myopathic form can be mild or may not occur at all leading to underdiagnosis [3]. The myopathic variant can manifest during childhood or early adulthood with exercise-induced myalgia and recurrent episodes of rhabdomyolysis (RM) of variable severity. The variant p.Ser113Leu represents about 70% of mutant alleles and is exclusively associated with the myopathic form [1].
We discuss here a challenging diagnosis of CPT II-D in a child after he presented with a severe first attack of exercise-induced RM. He carries the common p.Ser113Leu variant and a variant of unknown significance in the CPT II gene [4].
Case presentation
A 10-year-old boy presented to the emergency department with a one-day history of lower limb pain that progressed to an inability to bear weight one hour after playing 90 minutes of football. He also complained of lower abdominal pain and noticed cola-colored urine. He denied other urinary symptoms, fever, or recent trauma, and this was his first episode.
He is a product of a non-consanguineous marriage and has five healthy siblings. There is no history of any genetic diseases running in the family. The birth history, growth, and development were normal. On examination, the patient was vitally stable and afebrile. His muscular power was five out of five in the upper and lower limbs, but could not bear weight because of the pain. Otherwise, there was no muscular tenderness, and deep tendon reflexes and sensations were intact. Urine analysis showed red urine with a white blood cell count of more than 50/hpf, red blood cell count of 11-20 /hpf, and negative nitrites. Blood tests showed high serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST). Other liver parameters, complete blood count, C-reactive protein, renal function tests, electrolytes, and lactic acid were all normal (Table 1). The child was admitted initially under the impression of glomerulonephritis and liver derangement. The next day, creatine kinase (CK) came to be very high as well as creatinine kinase myocardial band (CK-MB) suggestive of RM (Table 1).
In the following days, CK, AST, and ALT transaminases kept rising, reaching 164000 U/L, 4840 U/L, and 981 U/L, respectively (Figure 1), hence a fatty acid oxidation defect was suspected. The child was then started on one-and-a-half maintenance intravenous fluids with dextrose 10% normal saline providing a glucose infusion rate of 5.5 mg/kg/min along with an alkalinization and L-carnitine supplementation. RM workup included urine toxicology, high-performance liquid chromatography-tandem mass spectrometry (TMS) on dried blood spots, and urine amino acids. The ECG and echocardiogram were normal.
Creatine kinase, alanine aminotransferase, and aspartate aminotransferase trends.
Subsequently, the child began to be active and pain-free. While the CK levels took three days to normalize, liver enzymes remained elevated for up to two weeks (Figure 1).
TMS was normal, urine amino acids showed elevated aspartic acid, threonine, serine, asparagine, glutamine, glycine, alanine, cystine, lysine, histidine, and arginine. Urine toxicology was negative. However, urine organic acids were not collected.
The AllNeuro (CentoGene, Rostock, Germany) panel has been performed and revealed compound heterozygosity for the pathogenic variant c.338C>T (p.Ser113Leu) and a variant of unknown significance (VUS) c.729_731del (p.Leu244del) in the CPT II gene. The VUS was described as an in-frame deletion of three bps in exon four, which causes the loss of residue Leu at position 244. Each mutation and its pathogenicity were validated by bioinformatics analysis, Sanger sequencing-based co-segregation testing, and computational assessment. Computational methods have demonstrated their association with disease.
A high carbohydrate, low-fat diet, daily medium chain triglycerides (MCT) oil supplementation, and carnitine were prescribed. The child was advised to avoid fasting for more than 12 hours and to take a snack and an extra MCT dose 30 minutes before exercise.
During his follow-up, the patient continued to experience intermittently some degree of leg pain after walking even though his CK levels were normal. He was found to have low vitamin D and was given vitamin D supplements.
In four years, the patient experienced three more episodes of RM due to non-compliance to dietary treatment, which required admission. Since then, he has avoided any intensive physical activity.
Discussion
RM is a clinical condition caused by skeletal muscle breakdown due to multiple etiologies. These causes include severe trauma, vigorous exercise, burns, electrical injury, seizure, electrolyte imbalances, drugs, infectious diseases, genetic neuromuscular disorders, and metabolic myopathies (MMs) [5].
With the release of muscular intracytoplasmic proteins, a risk of life-threatening complications may develop such as hyperkalemia, hypocalcemia, hypovolemia, acute kidney injury, compartment syndrome, and rarely disseminated intravascular coagulation. RM should be suspected in patients presenting with a triad of myalgia, weakness, and reddish-to-brown urine. Elevated serum CK levels over 1000 U/L indicate RM [6]. In our patient, CK level reached 700 x upper limit of normal, which could have exposed the child to life-threatening complications due to a delay in the management on admission. Despite displaying the full triad of RM, our patient was initially misdiagnosed as acute hepatitis because of markedly elevated liver enzymes; however, an AST-to-ALT ratio of > 2:1 is suggestive of muscle injury rather than hepatitis [7].
Inherited metabolic disorder (IMD) as a cause of RM is often missed; in 475 patients admitted for RM, 10% were related to underlying genetic or MM [8]. MM can be caused mainly by glycogen storage disorders, fatty acid oxidation defects (FAODs), or mitochondrial disorders. Other MMs are listed in Table 2 [5,7,9].
Physicians should have a high degree of suspicion of an underlying IMD if there is a personal or family history of recurrent RM or exercise intolerance (cramps, myalgia) [10]. It is important to note that the presence of an identifiable trigger does not necessarily exclude an underlying genetic cause [11]. As in our case, the patient denied any history of exercise intolerance before the first attack.
In long-chain fatty acid oxidation disorders (LCFAODs), attacks or myoglobinuria occur typically after mild to moderate prolonged exercise or when patients are additionally stressed by fasting, cold exposure, or infection. RM may present as either isolated or in association with life-threatening manifestations such as hypoglycemia, cardiomyopathy, arrhythmia, liver impairment, and/or encephalopathy [12].
The myopathic form of CPT II-D is the most common lipid metabolism disorder affecting skeletal muscle [10]. Even though the myopathic form is also called the adult form, 60% of patients manifested their symptoms below the age of 12 years [1].
In contrast to patients with other MMs, no residual weakness or persistent hyperCKemia is seen between the attacks in CPT II-D whereas only 10% of affected individuals have been observed with a consistent elevation of serum CK level [5,13].
The diagnosis of CPT II-D can be challenging. The acylcarnitines analysis performed on dried blood spots (DBS) for our patient in the first two attacks did not show the typical profile of elevated C16 and C18:2 species, and the increased C16+ C18:1/C2 ratio [14]. This test should be performed in plasma or serum as the diagnosis can be missed when it is performed in DBS [15]. Furthermore, normal plasma acylcarnitines do not rule out the diagnosis even during the acute attack. Urine organic acids might show non-specific dicarboxylic aciduria. The generalized aminoaciduria observed in our patient was related to muscle protein release.
FAOD study can be performed on fibroblasts or muscle tissue; however, molecular genetic testing has become the gold standard for establishing a definitive diagnosis [1].
Due to the broad clinical and genetic variability of FAOD, diagnosing them quickly and precisely is challenging. To date, 179 likely or pathogenic mutations have been reported in association with the myopathic form [16].
In the present case, compound heterozygous mutations have been found. The p.Ser113Leu variation accounts for approximately 70% of mutant alleles and is linked explicitly to the myopathic type [1,17]. The protein length changing c.729_731del (p.Leu244del) mutation belongs to evolutionary highly conserved regions. Silico tools showed that the mutation was disease-causing. This variant has been reported in one patient with the lethal neonatal form [4].
It is advised to evaluate even asymptomatic relatives to reduce the morbidity and mortality rate. This is particularly crucial before any general anesthesia, which might expose to severe RM [3].
Acute management consists of early fluid resuscitation with dextrose 10% at a rate of one and a half to double maintenance to enhance the anabolism and ensure sufficient urine output [12]. The use of carnitine is controversial during acute attacks of LCFAOD [18].
The dietary management of LCFAOD consists of a high carbohydrate intake covering 60% of the total energy requirement. Fat intake should be limited to 30-35%, with long-chain fat restricted to only 15% of the total energy. MCT supplementation covers the remainder of the energy requirement. Patients should avoid identifiable triggers, such as extended fasting and prolonged exercise. Moreover, a low-fat snack with MCT oil should be provided 30 to 45 minutes before strenuous exercise. Patients should also be monitored for secondary carnitine deficiency [12].
Conclusions
This case report highlights the importance of metabolic and genetic testing in patients presenting with RM. Although the clinical presentation of myopathic CPT II-D was typical, TMS on DBS was not diagnostic. The physician should inquire about personal or family history of exercise-induced myalgia or intolerance and collect critical samples before starting hyperhydration with high glucose concentration fluid. Genetic testing remains the gold standard diagnostic test. Our data offer novel perspectives for understanding the relationship between genotypes and phenotypes in CPT II-D. Further functional studies should be undertaken to investigate the effects of mutations on the disease.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Muscle carnitine palmitoyltransferase II (CPT II) deficiency: a conceptual approach Molecules Joshi PR Zierz S 17842520203229503710.3390/molecules 25081784 PMC 7221885 · doi ↗ · pubmed ↗
- 2Mitochondrial fatty acid β-oxidation disorders: from disease to lipidomic studies-a critical review Int J Mol Sci Guerra IM Ferreira HB Melo T 139332320223643041910.3390/ijms 232213933 PMC 9696092 · doi ↗ · pubmed ↗
- 3Carnitine palmitoyltransferase II deficiency Gene Reviews Wieser T Seattle, WA University of Washington 2024 https://www.ncbi.nlm.nih.gov/books/NBK 1253/
- 4A novel mutation leading to the lethal form of carnitine palmitoyltransferase type-2 deficiency J Pediatr Endocrinol Metab Dorum S Güney Varal I Gorukmez O Dogan P Ekici A 7817833220193119977410.1515/jpem-2019-0038 · doi ↗ · pubmed ↗
- 5A metabolism perspective on pediatric rhabdomyolysis Trend Pediat YazıcıH Kalkan Uçar S 14715322021
- 6Rhabdomyolysis: revisited Ulster Med J Gupta A Thorson P Penmatsa KR Gupta P 6169902021 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC 8278949/34276082 PMC 8278949 · pubmed ↗
- 7Carnitine palmitoyltransferase-II deficiency: case presentation and review of the literature Rom J Intern Med Mccormick BJ Chirila RM 4204245920213411880010.2478/rjim-2021-0021 · doi ↗ · pubmed ↗
- 8Rhabdomyolysis: an evaluation of 475 hospitalized patients Medicine (Baltimore) Melli G Chaudhry V Cornblath DR 3773858420051626741210.1097/01.md.0000188565.48918.41 · doi ↗ · pubmed ↗