Intact and mutated Shigella diguanylate cyclases increase c-di-GMP
Ruchi Ojha, Stefanie Krug, Prentiss Jones, Benjamin J. Koestler

TL;DR
This study shows that both intact and mutated DGC enzymes in Shigella contribute to c-di-GMP signaling, which is important for the bacterium's adaptation and pathogenesis.
Contribution
The study reveals that degenerate DGC pseudogenes in Shigella still produce c-di-GMP, challenging the assumption that mutations inactivate these enzymes.
Findings
Intact DGCs in Shigella synthesize c-di-GMP at varying levels in vitro and during infection.
Degenerate DGCs with nonsense mutations still produce c-di-GMP, suggesting functional contributions despite mutations.
Expression of certain DGCs affects invasion, plaque formation, and acid sensitivity, independent of c-di-GMP levels.
Abstract
The intracellular human pathogen Shigella invades the colonic epithelium to cause disease. Prior to invasion, this bacterium navigates through different environments within the human body, including the stomach and the small intestine. To adapt to changing environments, Shigella uses the bacterial second messenger cyclic di-GMP (c di-GMP) signaling system, synthesized by diguanylate cyclases (DGCs) encoding GGDEF domains. Shigella flexneri encodes a total of 9 GGDEF or GGDEF-EAL domain enzymes in its genome, but five of these genes have acquired mutations that presumably inactivated the c-di-GMP synthesis activity of these enzymes. In this study, we examined individual S. flexneri DGCs for their role in c-di-GMP synthesis and pathogenesis. We individually expressed each of the four intact DGCs in a S. flexneri strain, where these four DGCs had been deleted (Δ4DGC). We found that the 4…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1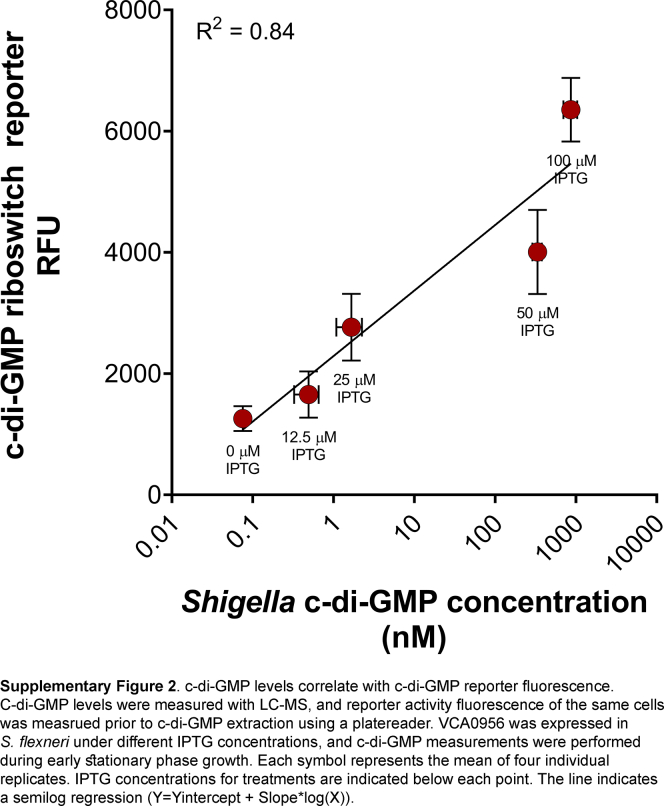 Figure 2
Figure 2 Figure 3
Figure 3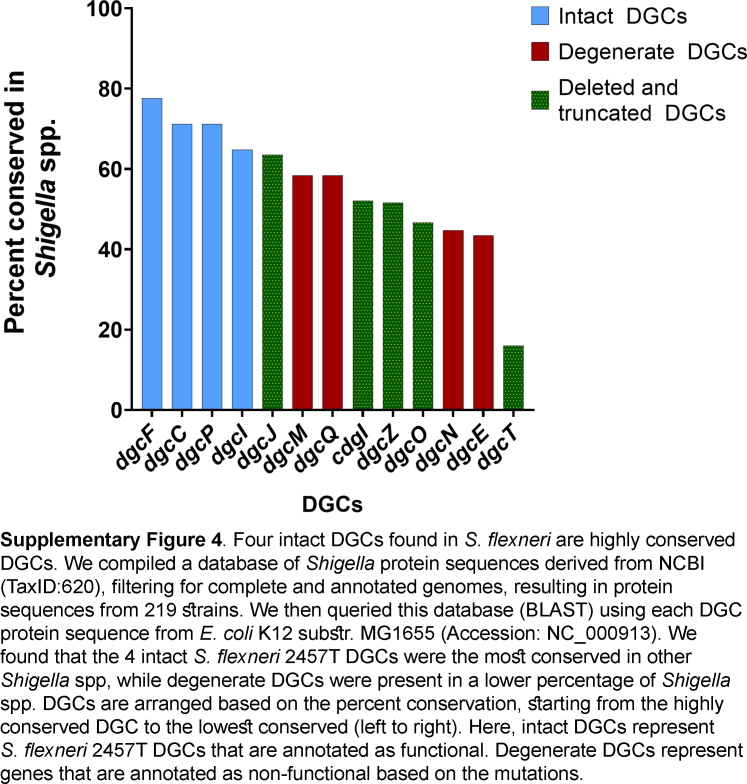 Figure 4
Figure 4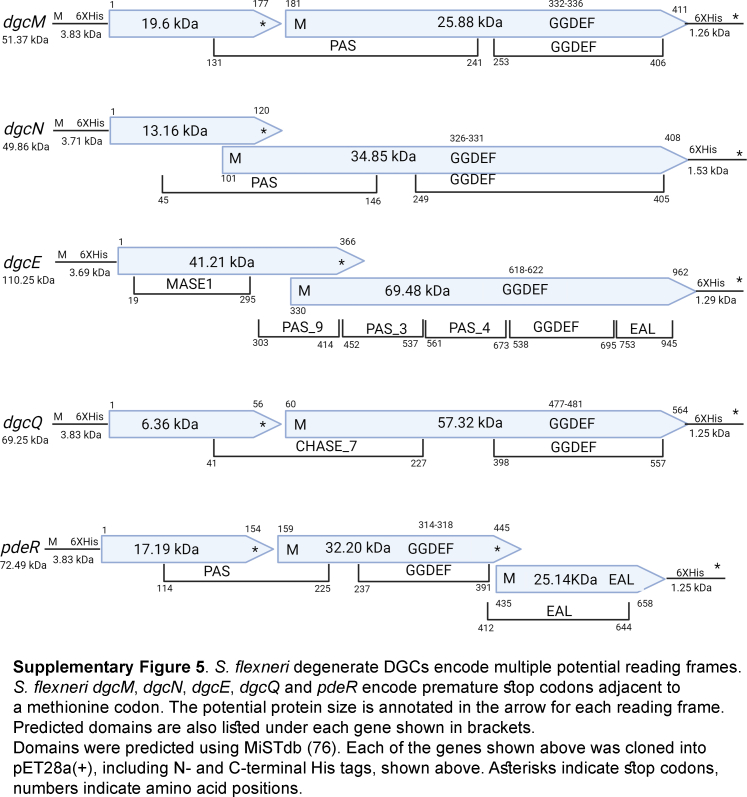 Figure 5
Figure 5 Figure 6
Figure 6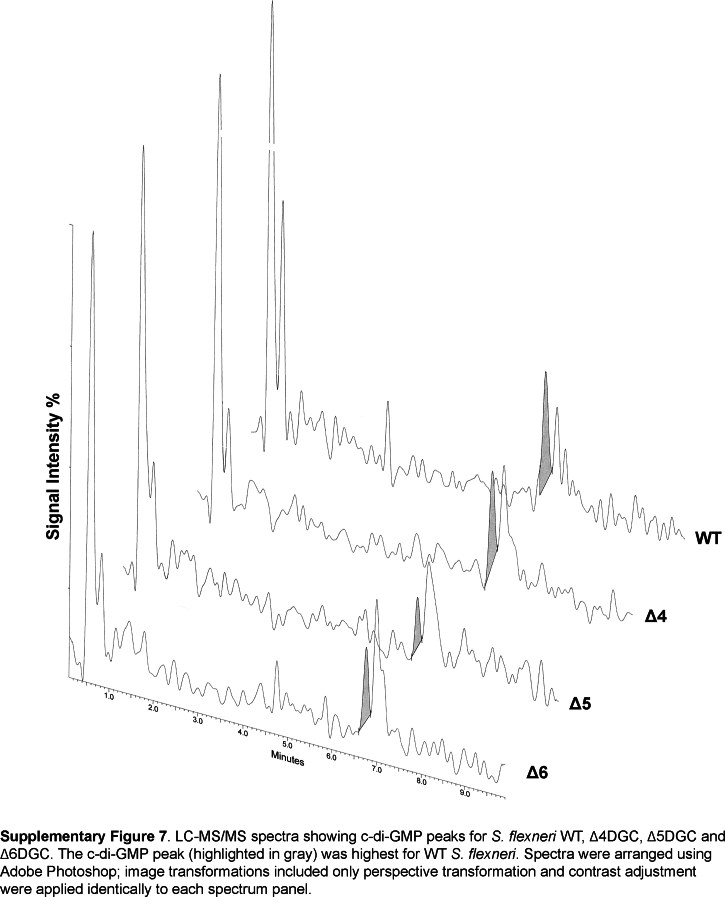 Figure 7
Figure 7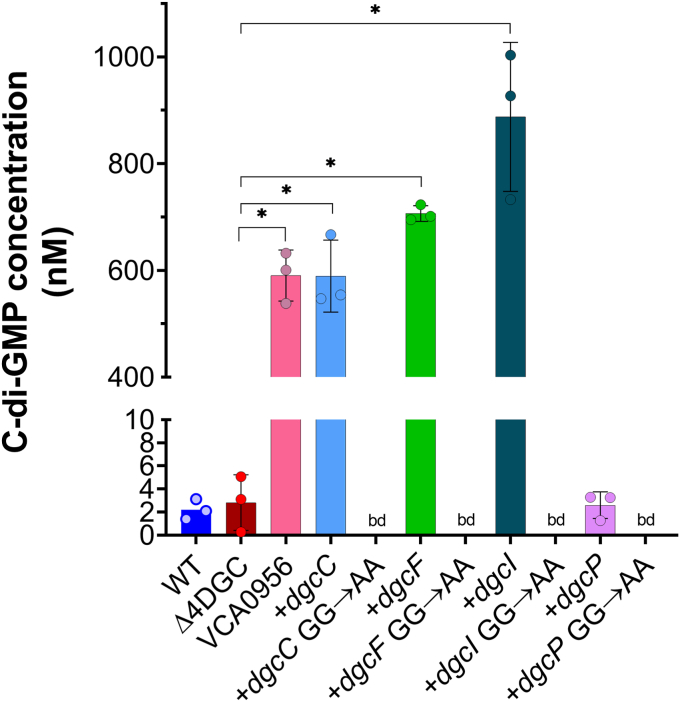 Figure 8
Figure 8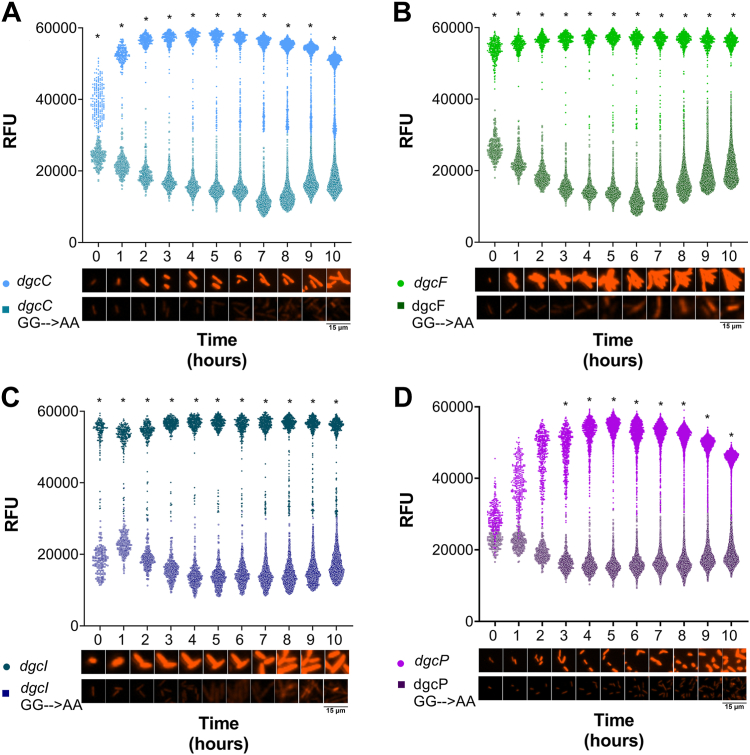 Figure 9
Figure 9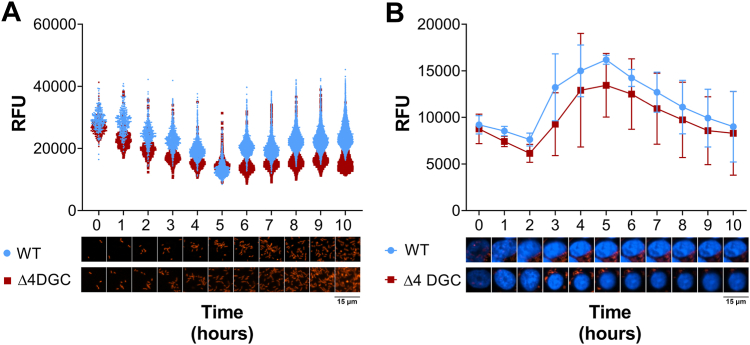 Figure 10
Figure 10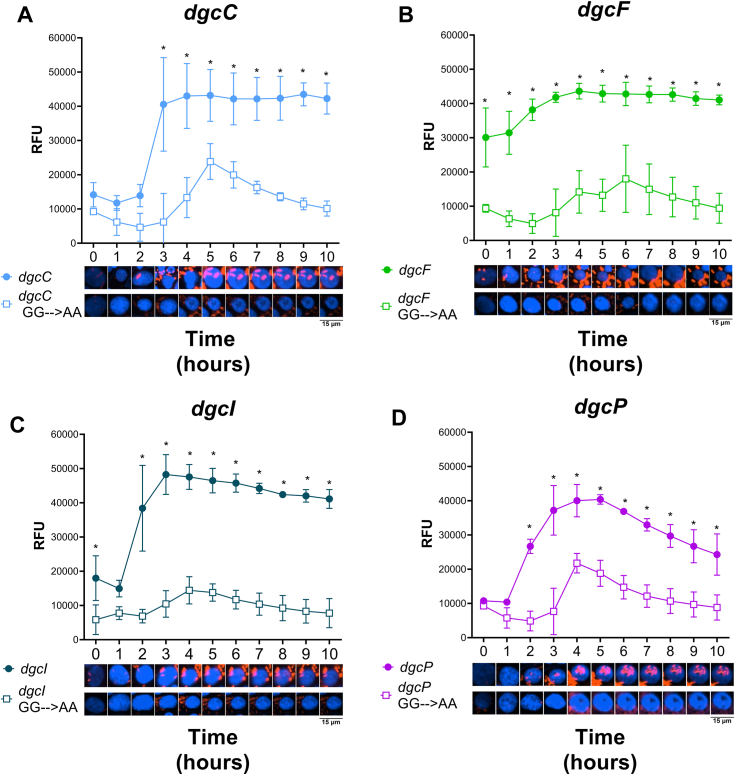 Figure 11
Figure 11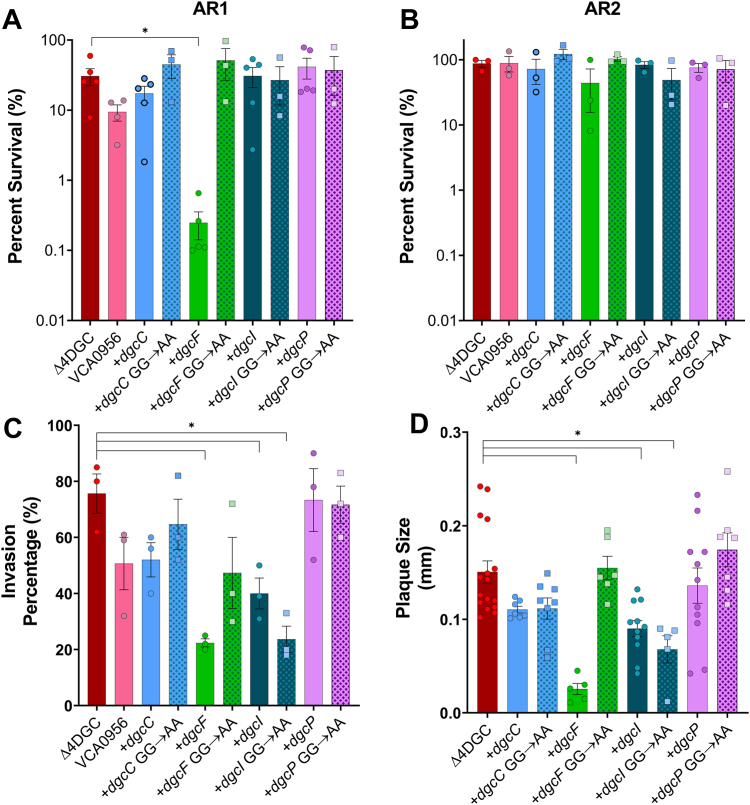 Figure 12
Figure 12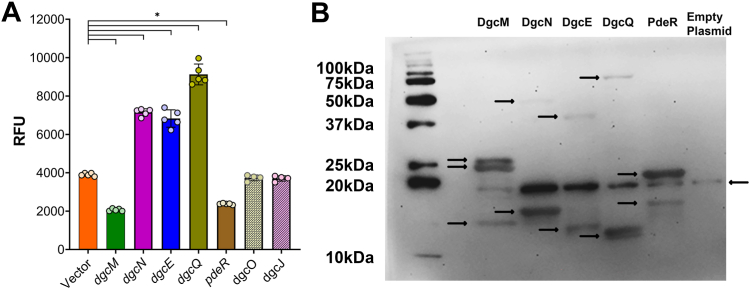 Figure 13
Figure 13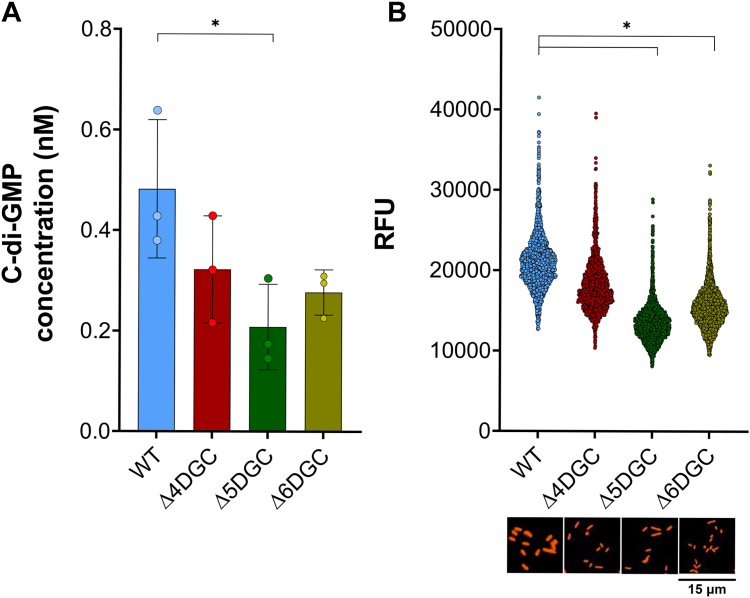 Figure 14
Figure 14Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEscherichia coli research studies · Bacterial Genetics and Biotechnology · CRISPR and Genetic Engineering
Shigellosis, caused by the human pathogen Shigella, is a prominent gastrointestinal infection in developing countries (1, 2). Shigella flexneri evolved from commensal Escherichia coli to efficiently infect the human gastrointestinal tract after navigating different host microenvironments (3, 4). With the global rise in antibiotic resistance among Shigella strains (5, 6), and the lack of vaccines for Shigellosis prevention (1, 7), it is imperative to understand Shigella pathogenesis mechanisms. In the colon, Shigella invades the epithelium using a type III secretion system encoded on a ∼220kb virulence plasmid (7, 8, 9, 10). Once inside colonic epithelial cells, Shigella multiplies and spreads using actin-based motility (11, 12, 13).
During infection, S. flexneri senses and responds to many different environmental signals to adapt to various microenvironments within the human body. For example, lower pH in the stomach alters the expression of acid-related genes and induces biofilm formation (14, 15), bile acids in the small intestine promote initial adhesion and invasion (16, 17, 18), and formate within a host cell promotes cell-to-cell spread (19). Bacteria like S. flexneri encode many different systems to sense and respond to these signals, one of which is the second messenger cyclic di-GMP (c di-GMP) signaling system (20, 21).
C-di-GMP signaling is widely conserved in most bacteria (22, 23) and regulates diverse phenotypes (24, 25, 26, 27, 28). C-di-GMP synthesis is driven by diguanylate cyclases (DGCs) encoding a C-terminal GGDEF domain, which brings together two GTP molecules. Alterations of the GGDEF catalytic site can eliminate c-di-GMP synthesis of these enzymes (29, 30, 31). Conversely, c-di-GMP is degraded by c-di-GMP–specific phosphodiesterases encoding an EAL domain (22, 23, 28). In many bacteria, c-di-GMP promotes biofilm formation and reduces invasive capacity (7, 20, 32). However, certain DGCs and phosphodiesterases synthesize local pools of c-di-GMP, allowing them to regulate specific phenotypes regardless of overall c-di-GMP levels; this phenomenon is known as signaling specificity (31, 33, 34). E. coli K12 encodes 19 GGDEF-containing genes in its genome (35); in comparison, S. flexneri encodes nine GGDEF or GGDEF-EAL–containing genes, but the majority of these genes contain mutations that presumably eliminate c-di-GMP synthesis activity (pseudogenes) (36). S. flexneri has four DGCs predicted to synthesize c-di-GMP: dgcC, dgcF, dgcI, and dgcP (36).
We previously showed that deletion of intact S. flexneri DGCs alters pathogenesis-related phenotypes (20). Here, we investigate how S. flexneri DGCs contribute to c-di-GMP levels and how the expression of these genes impacts different phenotypes. To this end, we created a mutant S. flexneri strain where all four intact DGCs were deleted (Δ4DGC) and then expressed each S. flexneri DGC and respective active site mutant DGCs (GG→AA) from plasmids. We observed that all four DGCs were able to synthesize c-di-GMP at different levels and that each gene differentially contributed to acid resistance (AR), invasion, and plaque formation. Interestingly, we still detected c-di-GMP in our S. flexneri Δ4DGC strain, which led us to examine the five putative S. flexneri DGC pseudogenes. We found that expression of S. flexneri dgcE, dgcQ, and dgcN, each of which have nonsense mutations prior to the GGDEF domain, increased c-di-GMP, and deletion of S. flexneri ΔdgcE (Δ5DGC) and ΔdgcEΔdgcQ (Δ6DGC) from the Δ4DGC strain significantly reduced c-di-GMP. This provides evidence that S. flexneri pseudogenes can retain function.
Results
S. flexneri DGCs increase c-di-GMP
The S. flexneri 2457T genome encodes four intact GGDEF domain enzymes, dgcC (S0329), dgcF (S1698), dgcI (S0827), and dgcP (S1545) (36). Individual deletion of DGCs from the S. flexneri genome significantly reduces biofilm formation and alters invasion, plaque formation, and AR in a DGC-specific manner (20). To study how individual S. flexneri DGCs contribute to pathogenesis, we generated a mutant strain where all four intact DGCs (dgcC, dgcF, dgcI, and dgcP) were deleted (Δ4DGC). Each of these four S. flexneri DGCs were then individually expressed from an IPTG-inducible plasmid in the S. flexneri Δ4DGC background. As a negative control, we mutated two amino acids from the active site of each of the four S. flexneri DGCs from GG(D/E)EF to AA(D/E)EF (glycine to alanine; GG→AA) to separate the regulatory role of the protein itself to its c-di-GMP synthesis (29). This mutation has been shown to eliminate c-di-GMP synthesis in DGCs from other bacteria (29, 30, 37). We confirmed that the expression of these four DGCs did not alter S. flexneri growth in broth (Fig. S1).
Ectopic expression of the Vibrio cholerae DGC VCA0956 significantly increases c-di-GMP in S. flexneri as measured by LC-MS/MS (20). We hypothesized that the expression of intact S. flexneri DGCs will increase c-di-GMP levels as compared to GG→AA mutant alleles. C-di-GMP from these strains was extracted during mid log growth (∼3 h post IPTG addition) and quantified using LC-MS/MS. We observed that dgcI expression resulted in the highest levels of c-di-GMP, whereas dgcC, and dgcF expression increased c-di-GMP to levels comparable to the VCA0956 expression strain. Contrary to that, the dgcP expression strain showed no difference in c-di-GMP levels to that of the Δ4DGC strain (Fig. 1). C-di-GMP was below our limit of detection when individual GG→AA DGC mutants were expressed, but interestingly, we observed detectable levels of c-di-GMP in the S. flexneri Δ4DGC strain carrying an empty plasmid, comparable to the WT strain (Fig. 1).Figure 1Shigella flexneri intact DGCs regulate c-di-GMP levels. Individual S. flexneri DGCs were expressed in the S. flexneri Δ4DGC strain, and c-di-GMP levels were measured using LC-MS/MS. dgcC, dgcF, and dgcI expression in the Δ4DGC strain showed significantly more c-di-GMP synthesis than the GG→AA mutants or the empty vector controls, extracted at mid log phase. Expression of dgcP did not significantly alter S. flexneri c-di-GMP levels in comparison to the Δ4DGC strain. We also noticed no significant difference between WT S. flexneri and the Δ4DGC strain. VCA0956 (positive control) significantly increased c-di-GMP levels in S. flexneri (20). bd indicates below detection, ∗ represents significant differences between strains as analyzed by one-way ANOVA with Sidak’s multiple comparisons post test (p < 0.05). Each symbol represents independent replicates, and error bars indicate SD. c di-GMP, cyclic di-GMP; DGC, diguanylate cyclase.
S. flexneri dgcP expression increases c-di-GMP 3 h postinduction
We were intrigued that S. flexneri dgcP expression did not increase c-di-GMP levels compared to the other three DGCs (Fig. 1). We further wanted to investigate temporal dynamics of the four intact S. flexneri DGCs; thus, as a complementary approach, we quantified relative c-di-GMP levels using a plasmid encoding a double tandem riboswitch Bc3-Bc4 controlling the expression of the reporter gene m-Scarlett I (riboswitch reporter)(38), allowing c-di-GMP measurement with single-cell resolution of live cells by microscopy. The riboswitch reporter binds c-di-GMP with high affinity and specificity, and in the absence of c-di-GMP, a terminator inhibits the translation of m-Scarlett I. This c-di-GMP reporter was validated in the closely related E. coli (38). We confirmed that the reporter functions similarly in S. flexneri, by comparing our positive control, VCA0956 LC-MS/MS measurements to the c-di-GMP reporter fluorescence from the same bacterial cultures (Fig. S2). We hypothesized that S. flexneri DGC expression would result in m-Scarlett I fluorescence that correlates with c-di-GMP levels measured by LC-MS/MS. We quantified m-Scarlett I fluorescence of our S. flexneri DGC expression strains over time using live-cell fluorescence microscopy. We then compared the mean of individual fluorescent cells of each DGC to the GG→AA mutants. Of note, we observed no differences in cell size and growth among all our strains. As expected, c-di-GMP–dependent fluorescence increased rapidly and significantly over time in S. flexneri dgcC, dgcF, and dgcI, reaching near maximal fluorescence after 1 h (Fig. 2, A–C). Interestingly, S. flexneri dgcP expression increased fluorescence at a slower rate, resulting in significantly higher c-di-GMP levels 3 h post IPTG induction than the GG→AA dgcP mutant (Fig. 2D). We observed similar patterns when the fluorescence of these strains was quantified using a plate reader (Fig. S3). We speculate that differences in the growth conditions used for quantitation by LC-MS/MS and microscopy alter S. flexneri DgcP c-di-GMP synthesis (Figs. 1 and 2D).Figure 2DGC expression significantly increases c-di-GMP–dependent fluorescence compared to GG→AA mutants. Single bacterial cell fluorescence (relative fluorescence units, RFU) was measured using microscopy over time for the Shigella flexneri Δ4DGC strain expressing (A) dgcC, (B) dgcF, (C) dgcI, and (D) dgcP (lighter colored circles), alongside their respective GG→AA mutants (darker colored*, open**squares*). S. flexneri DGCs were expressed from an IPTG-inducible plasmid and a second plasmid expressing the c-di-GMP–specific double tandem riboswitch with m-Scarlett I (riboswitch reporter) was used to measure c-di-GMP–dependent fluorescence. Each of the four DGC expression strains fluoresces more than the GG→AA mutant expression strains. Symbols indicate individual fluorescent cells for each time point for each strain. The graph is representative of one of the three independent trials. Images below the graph (A–D) are representative samples of individual bacterial cells; red indicates c-di-GMP–dependent m-Scarlett I fluorescence. Significant fluorescence differences between strains were analyzed using two-way ANOVA with Sidak’s multiple comparisons post test (∗p < 0.05) at each time point. c di-GMP, cyclic di-GMP; DGC, diguanylate cyclase.
S. flexneri c-di-GMP levels are dynamic during host cell growth
As S. flexneri is an intracellular pathogen, we were interested in c-di-GMP changes during growth in a host cell, and if the four intact S. flexneri DGCs are responsible for c-di-GMP production in this environment. To answer these questions, we used our riboswitch reporter to evaluate changes in c-di-GMP production of live S. flexneri cells over time. When WT S. flexneri was grown in a defined medium, c-di-GMP remained relatively constant, exhibiting modestly decreasing fluorescence over time, although this trend was not statistically significant. The S. flexneri Δ4DGC strain was consistently lower than the WT at all time points, although this difference too was not statistically significant (Fig. 3A).Figure 3Shigella flexneri increases c-di-GMP production in vivo 3 h post infection. c-di-GMP–driven fluorescence (RFU) of S. flexneri WT and the Δ4DGC strain carrying the c-di-GMP riboswitch reporter grown (A) in culture or (B) in Henle-407 cells. A, in culture, c-di-GMP reporter fluorescence levels remained relatively constant over the course of 10 h and were consistently but nonsignificantly higher in the WT S. flexneri strain than the Δ4DGC strain. Symbols indicate the individual fluorescent cells, representative of one trial among the three. B, in both the S. flexneri WT and Δ4DGC strain, c-di-GMP reporter fluorescence levels increased 3 HPI and then gradually decreased starting at 5 HPI. Images below the graph are representative of individual bacterial cells in culture. Infected Henle-407 cells were stained with Hoechst to label host cell nuclei (blue). Red indicates c-di-GMP–dependent m-Scarlett I fluorescence. Significant differences between strains were analyzed using two-way ANOVA with Sidak’s multiple comparisons posttest (∗p < 0.05). c di-GMP, cyclic di-GMP; DGC, diguanylate cyclase; RFU, relative fluorescence unit.
We performed the same analysis to assess changing c-di-GMP reporter fluorescence over time during S. flexneri infection of Henle-407 cells (Fig. 3B)(39). In contrast to S. flexneri grown in culture, WT S. flexneri fluorescence increased at 3 h postinfection (HPI), peaked 5 HPI, and then gradually decreased fluorescence during Henle-407 infection. For the S. flexneri Δ4DGC strain, we again observed that c-di-GMP reporter fluorescence levels were consistently nonsignificantly lower than in the WT strain, but we saw the same general pattern, where fluorescence increased at 3 HPI, peaked at 5 HPI, and then decreased afterward (Fig. 3B).
S. flexneri DGCs produce c-di-GMP during host cell infection
To determine how S. flexneri DGCs contribute to changing c-di-GMP levels during host cell growth, we compared the expression of S. flexneri dgcC, dgcF, dgcI, dgcP, and their respective GG→AA mutants in the S. flexneri Δ4DGC strain during infection. Expression of each of the four intact DGCs increased c-di-GMP reporter fluorescence, but the temporal dynamics were different for each strain (Fig. 4). Specifically, the time that it took each DGC to initiate c-di-GMP synthesis varied. dgcC expression exhibited no significant fluorescence until 3 HPI and then increased rapidly (Fig. 4A). dgcF expression resulted in constitutively high c-di-GMP reporter fluorescence throughout the entire course of infection (Fig. 4B). dgcI expression did not increase fluorescence until 2 HPI (Fig. 4C). Similar to dgcI, dgcP expression increased starting at 2 HPI and peaked at 5 HPI and then steadily decreased for the remainder of the experiment (Fig. 4D). Interestingly, for each of our GG→AA mutants, we observed an increase in fluorescence that peaked between 4 and 6 HPI and then decreased as the infection progressed.Figure 4Shigella flexneri DGCs regulate c-di-GMP synthesis post infection. C-di-GMP–dependent fluorescence (RFU) of the S. flexneri Δ4DGC strain expressing (A) dgcC, (B) dgcF, (C) dgcI, or (D) dgcP, alongside their respective GG→AA mutant alleles, was quantified during infection of Henle-407 cells. Expression of all four intact DGCs significantly increased fluorescence compared to individual GG→AA mutant DGCs, but the time it took for these differences to become significant was variable. Representative micrographs below illustrate single-cell fluorescence; red indicates c-di-GMP–dependent m-Scarlett I fluorescence, and blue host cell nuclei stained with Hoechst. Symbols indicate the mean fluorescence of single-cell measurements from three independent replicates, and error bars indicate SD. Significant differences between strains at each time point were analyzed using two-way ANOVA with Sidak’s multiple comparisons posttest (∗p < 0.05). c di-GMP, cyclic di-GMP; DGC, diguanylate cyclase; RFU, relative fluorescence unit.
dgcF expression increases S. flexneri acid sensitivity in AR1-inducing conditions
During human infection, Shigella must survive the highly acidic environment in the stomach before it can access the human colon. Shigella survives acid stress with at least two systems (AR1 and AR2) that are induced in different conditions (40, 41, 42, 43, 44). We previously showed that deletion of S. flexneri dgcF increases AR (20); therefore, we hypothesized that expression of S. flexneri dgcF will decrease AR. We preconditioned S. flexneri DGCs in complex media at pH 5.5 to induce AR1 and separately at pH 5.5 with glucose and low oxygen to induce AR2. We then challenged our DGC expression strains with exposure to defined media at pH 2.5 (to study AR1) or pH 2.5 with glutamate (to study AR2) (40, 41, 42, 45). dgcF expression significantly reduced acid survival in AR1-inducing conditions, compared to other DGCs and the GG→AA mutants (Fig. 5A). In AR2-inducing conditions, DGC expression did not significantly decrease AR (Fig. 5B). This suggests that S. flexneri dgcF specifically regulates acid survival in AR1-inducing conditions.Figure 5**Expression of Shigella flexneri dgcF increases acid sensitivity in AR1-inducing conditions and reduces virulence.**A, each S. flexneri intact DGC was expressed in the Δ4DGC strain and challenged in pH 2.5 medium in AR1-inducing conditions for 1 h. Percent survival was determined by quantifying the subsequent CFU/ml of cultures challenged in pH 2.5 medium, compared to organisms in pH 7.0 medium. dgcF expression significantly decreased acid resistance as compared to the S. flexneri Δ4DGC strain carrying an empty plasmid and the GG→AA mutant dgcF, as determined by a Kruskal–Wallis test with Dunn’s multiple comparisons post test. B, the same experiment was performed in AR2-inducing conditions, which relies on external glutamate (41, 42). AR2-inducing conditions resulted in overall higher S. flexneri survival, and we observed no significant differences in survival when each intact DGC or GG→AA mutant was expressed. Bars represent the mean of three independent trials, and error represents SD. C, we assessed the capacity of S. flexneri strains to invade Henle-407 cells. dgcF, dgcI, and the dgcI GG→AA mutant expression in the Δ4DGC strain show significant reduction in the percentage of invaded cells. Bars indicate the mean of three independent trials, and error represents SD. D, plaque size was measured after infecting Henle-407 cells with the S. flexneri Δ4DGC strain expressing the four intact DGCs or GG→AA mutant alleles. dgcF, dgcI, and the GG→AA mutant dgcI strains show significant reduction in plaque size as compared to the S. flexneri Δ4DGC mutant strain carrying an empty plasmid. Symbols represent individual plaques from one replicate among the three individual trials. Bars indicate the mean, and error bars represents the SD. Significant differences were analyzed using two-way ANOVA with Sidak’s multiple comparisons posttest (∗p < 0.05). AR, acid resistance; c di-GMP, cyclic di-GMP; DGC, diguanylate cyclase.
Virulence phenotypes in S. flexneri are regulated by dgcF and dgcI
VCA0956 expression reduces S. flexneri invasion and plaque formation, but the S. flexneri ΔdgcF strain also has reduced invasion and forms smaller plaques in Henle-407 monolayers (20). To see how expressing the four intact S. flexneri DGCs impacts these phenotypes, we examined the invasion and plaque phenotypes affected by our DGC expression strains. Henle-407 cells were infected with the S. flexneri Δ4DGC strain with DGC expression plasmids and their respective GG→AA mutants. As a control, we included VCA0956 expression which decreases invasion (20). Expression of dgcF and dgcI exhibited invasion defects as compared to the empty plasmid control, while dgcC and dgcP expression strains show no significant difference in invasion (Fig. 5C). Interestingly, the dgcI GG→AA mutant also exhibited an invasion defect, while all other GG→AA DGC mutants did not.
We also examined the ability of each DGC expression strain to form plaques in cell culture monolayers, as previously described (39). Each strain was induced 30 min postinfection to ensure comparable invasion. Similar to the invasion assay, we found that the dgcF, dgcI, and dgcI GG→AA mutant showed reduced plaque size in comparison to the empty plasmid control, while dgcC and dgcP expression showed no significant difference in plaque size compared to the Δ4DGC (Fig. 5D).
Four S. flexneri DGC pseudogenes encode GGDEF domains after nonsense mutations
Our LC-MS/MS measurements and riboswitch reporter data indicate that the S. flexneri Δ4DGC strain still produces c-di-GMP; therefore, we sought to identify other S. flexneri genes that synthesize c-di-GMP. We started by investigating the other DGC pseudogenes. In comparison to E.coli K12 substr. MG1655 (Accession: NC_000913), four GGDEF domain and one GGDEF-EAL domain genes are annotated as pseudogenes (considered degenerate) in S. flexneri 2457T (Accession: AE014073.1) and include deletions, frameshift mutations, and/or nonsense mutations that presumably disrupt the GGDEF domain (20, 36). Compared to E. coli, there are nine other DGCs that either do not contain the GGDEF domain or are deleted in S. flexneri (36). Therefore, for the scope of this study, we focused on the 5 GGDEF and GGDEF-EAL domain encoding genes with premature stop codons (Table 1). Recent studies indicate that other S. flexneri degenerate enzymes have demonstrable phenotypes (46); therefore, we hypothesized that one or more of these DGC pseudogenes retain c-di-GMP synthesis. Compared to E. coli MG1655, dgcT, dgcZ and cdgI genes are completely absent from the S. flexneri 2457T genome (Table 1). S. flexneri dgcO (S1870) and dgcJ (S1553) genes have truncating deletions that remove the GGDEF domain. The remaining four S. flexneri genes, dgcM (S1445), dgcN (S2841), dgcE (S2255), and dgcQ (S2095), have pairwise identity >97.9% compared to E. coli K12 strain; however, each of these genes encodes a nonsense mutation resulting in a stop codon preceding the GGDEF domain, which we confirmed by whole genome sequencing. Notably, these four DGC genes have alternative reading frames that begin close to the nonsense mutation site and encode an intact GG[D/E]EF domain. S. flexneri dgcM, dgcE, and dgcQ all contain an RXXD inhibition site, potentially enabling allosteric feedback inhibition (Table 1) (47, 48). Interestingly, while the four intact S. flexneri 2475T DGCs are the most well conserved in other Shigella spp., degenerate DGCs are intact in the genomes of other Shigella spp. (Fig. S4).Table 1Shigella degenerate and deleted DGCsGeneLocusOther namesGGDEF domainMutationPredicted sizeI-siteA-sitedgcES2255yegERSSDVLARLGGDEFPremature Stop codon at 366 AA; MET at 33046.37 kDa and 70.79 kDadgcJS1553yeaJTruncating deletion, no GGDEFdgcMS1445ydaMRKGDLVFRWGGEEFPremature Stop codon at 177 AA; MET at 18023.88 kDa and 27.16 kDadgcNS2841yfiNGLRHKAYRLGGDEFPremature Stop codon at 120 AA; MET at 10117.23 kDa and 36.4 kDadgcOS1870yddV, dosCTruncating deletion, no GGDEFdgcQS2095yedQRAQDVAGRVGGEEFPremature Stop codon at 56 AA; MET at 5910.19 kDa and 58.7 kDadgcT**ycdTDeletiondgcZ**ydeHDeletioncdgI**yeaIDeletionpdeAS2600yfeAQENEKLYQLPGSELpdeFS2699yfgFEPGEDVYQLSGNDLpdeKS4206yhjKSPRMILAQISGYDFpdeOS1871yddU, dosPKPDQYLCRIEGTQFpdeRS1372yciREHDQVLARPGGDEFPremature Stop codon at 154 AA; MET at 16121.01 kDa, 32.20 kDa and 26.46 kDaShigella flexneri 2457T encodes 14 degenerate DGCs containing mutations that disrupt the GGDEF domain. To retain predicted function, the gene should encode GG[D/E]EF in the active site (A-site). The presence of RXXD (underlined) in the I-site indicates allosteric regulation. Five genes encode premature stop codons preceding the GGDEF domain, five genes include deletions that have removed the GGDEF domain, and four genes encode GGDEF domains without A-site residues required for c-di-GMP synthesis. Mutations were confirmed by whole-genome sequencing (Plasmidsaurus). The predicted protein size of genes encoding a premature stop and an alternative reading frame include a 6× His-tag.DGC, diguanylate cyclase.
S. flexneri dgcE and dgcQ produce c-di-GMP
To determine if S. flexneri degenerate DGCs are capable of c-di-GMP synthesis, we focused on the five degenerate DGCs that contain nonsense mutations preceding the GGDEF domain. We also included dgcO and dgcJ, with the expectation that these genes could not produce c-di-GMP due to the complete lack of GGDEF domain sequence. We expressed each of these 7 S. flexneri genes (the start codon to the orthologous E. coli stop codon) from a high expression plasmid, pet28a(+), with both N- and C-terminal 6X-His-tags in E. coli BL21 (Fig. S5). We chose this expression platform to increase the likelihood of observing potentially small amounts of c-di-GMP production from these gene products. We assessed c-di-GMP reporter fluorescence levels using our c-di-GMP riboswitch reporter (Fig. 6A). Surprisingly, dgcE, dgcQ, and dgcN expression resulted in fluorescence significantly higher than the empty plasmid control and other degenerate DGCs at 5 h post IPTG induction, consistent with c-di-GMP synthesis (Fig. 6A). dgcM and pdeR expression reduced fluorescence compared to the empty vector control. Expectedly, dgcO and dgcJ expression did not result in any increase in fluorescence relative to the empty plasmid control. Of note, we observed a modest reduction in growth upon dgcE and dgcN expression (Fig. S6). This growth inhibition could be due to stress associated with high expression of these gene products or from c-di-GMP production, as dgcN expression inhibits E. coli growth dependent on c-di-GMP synthesis (49, 50).Figure 6**Shigella flexneri dgcN, dgcE, and dgcQ significantly increase c-di-GMP levels.**A, S. flexneri degenerate DGCs were expressed in Escherichia coli BL21 and c-di-GMP levels measured using the c-di-GMP riboswitch reporter. dgcE, dgcQ, and dgcN had significantly increased fluorescence in comparison to our empty plasmid control, whereas dgcM and pdeR showed significantly lower fluorescence. Individual symbols represent three independent replicates; ∗ represents significant differences in comparison to the empty plasmid control as determined by one-way ANOVA with Sidak’s multiple comparisons posttest (p < 0.05). Error bars indicate SD. B, S. flexneri degenerate DGCs with both N- and C-terminal 6× His-tags were expressed in E. coli BL21, and protein expression was visualized by SDS-PAGE followed by Western blot. Samples for DgcN and DgcE were loaded 2× to visualize faint bands. Arrows highlight fragments for each gene expressed. We observed a nonspecific band at ∼20 kDa in all lanes including our empty vector control. c di-GMP, cyclic di-GMP; DGC, diguanylate cyclase.
We next assessed the protein expression of these five degenerate DGCs by Western blot, using the same plasmids with both N- and C-terminal 6× His-tags. Of note, a nonspecific band was observed at ∼20 kDa in all samples including our empty vector control (Fig. 6B). dgcM and dgcQ expression produced two proteins each, corresponding to their premature stop codon and adjacent methionine (Fig. 6B), presumably translating the GGDEF domain in second reading frame (Table 1). dgcM expression also produced a third band at ∼15 kDa, which does not match any reading frames. dgcN expression produced two bands at ∼17 kDa and ∼40 kDa. The 17 kDa size corresponds to the first N-terminal reading frame; however, the 40 kDa size corresponds with full-length DgcN. dgcE expression produced two bands at ∼45 kDa and 15 kDa. The 45 kDa corresponds to the N-terminal reading frame; however, the other band does not correspond with any potential dgcE reading frames. E. coli DgcE undergoes posttranslational proteolysis, thus it is possible that this processing is why the sizes we observed here did not match the predicted sizes (34, 51). pdeR expression also produced two bands; the first band at ∼18 kDa corresponds to the first reading frame, however the second band at ∼22 kDa does not match any predicted reading frames.
dgcE deletion reduces S. flexneri c-di-GMP levels
As expression of S. flexneri dgcE, dgcQ, and dgcN pseudogenes in E. coli BL-21 significantly increased c-di-GMP levels, we hypothesized that degenerate DGCs also contribute to c-di-GMP production in S. flexneri. To test this hypothesis, we first deleted dgcE from the S. flexneri Δ4DGC background and generated a Δ5DGC strain (Δ4DGC + ΔdgcE). Then, we deleted dgcQ in the S. flexneri Δ5DGC background, to generate Δ6DGC (Δ4DGC + ΔdgcE + ΔdgcQ). We quantified the c-di-GMP levels of each of these strains using LC-MS/MS (Fig. 7A). The S. flexneri Δ5DGC strain exhibited significantly reduced c-di-GMP levels compared to the WT S. flexneri strain; however, our Δ6DGC strain showed no significant difference compared to WT S. flexneri (Fig. 7A). Notably, we still detected a c-di-GMP peak in the S. flexneri Δ6DGC strain, indicating that dgcN (or other DGCs) may also produce c-di-GMP (Fig. S7). We confirmed our findings using the riboswitch reporter and fluorescence microscopy. Measurements using the riboswitch reporter mirrored those taken by LC-MS/MS, and we observed significant reductions in c-di-GMP reporter activity in both the Δ5DGC and Δ6DGC strains compared to the WT S. flexneri strain (Fig. 7B).Figure 7**Shigella flexneri Δ5DGC and Δ6DGC strains have significantly reduced c-di-GMP levels.**A, S. flexneri Δ5DGC (Δ4DGC + ΔdgcE) and Δ6DGC (Δ4DGC + ΔdgcE + ΔdgcQ) c-di-GMP levels were compared to WT S. flexneri using LC-MS/MS. The S. flexneri Δ5DGC strain showed significantly lower c-di-GMP levels than the WT S. flexneri strain. In the S. flexneri Δ6DGC strain, c-di-GMP levels were reduced but not statistically significant than in the WT strain. B, S. flexneri Δ5DGC and Δ6DGC strain c-di-GMP levels quantified using the c-di-GMP riboswitch reporter. C-di-GMP levels were significantly lower for the Δ5DGC or Δ6DGC strain than WT S. flexneri. Each symbol represents individual fluorescent cells for each strain. Images below the graph are individual fluorescent cells for each strain. Asterisks represent significant differences among strains as compared using one-way ANOVA with Sidak’s multiple comparisons post test (p < 0.05), error bars indicate SD. c di-GMP, cyclic di-GMP; DGC, diguanylate cyclase.
Discussion
Because of the evolutionary relationship between commensal E. coli and pathogenic S. flexneri, we can use these two organisms as a model to examine how c-di-GMP signaling changes in the process of pathoadaptation (52). Only the four DGCs most conserved in other Shigella spp. (Fig. S4) have been retained with high fidelity in S. flexneri 2457T. One possible reason that these enzymes were conserved over others is signaling specificity, where specific DGCs alter c-di-GMP synthesis and associated phenotypes (31, 33, 34). Our data support this hypothesis, as there is a poor correlation between the expression of each of the four intact S. flexneri DGCs and the phenotypes we measured in this study. For example, dgcF expression produces less c-di-GMP than dgcI expression in S. flexneri, but it is the only DGC that significantly decreases acid sensitivity in AR1-inducing conditions. Our observation that S. flexneri GG→AA mutated dgcI regulated invasion and plaque phenotypes irrespective of catalytic activity is also suggestive that protein–protein interactions are enabling specific phenotypes (24, 53, 54). An alternative explanation regarding the discrepancy between measured c-di-GMP and various phenotypes resulting from DGC expression is that environmental regulation is contributing to differences we observe between c-di-GMP measurements and phenotypes, as environmental conditions in phenotypic assays do not precisely match those used for c-di-GMP measurements, and DGCs recognize environmental signals via their variable N-terminal domains that alter enzymatic activity at the C-terminal domain (33, 55). One limitation of this study is that although these genes are being expressed from the same promoter in these experiments, it is also possible that differences in protein expression could be contributing to c-di-GMP–associated differences we observe here.
We observed that c-di-GMP reporter fluorescence levels inside host cells were dynamic during infection (Fig. 3), and that S. flexneri DGCs produced c-di-GMP differentially in this environment (Fig. 4). S. flexneri virulence gene expression is dynamic during growth in an epithelial cell, and the timing correlates with the increase in c-di-GMP we observed (10, 53, 56, 57, 58). Expression of VCA0956 in S. flexneri, which raises c-di-GMP, significantly downregulates the expression of virulence genes, including those of the type III secretion system (20), and a similar phenomenon was also reported in Salmonella typhimurium (24). Thus, it is possible that c-di-GMP is contributing to differential gene expression during growth in a host cell, but the signal transduction pathway linking c-di-GMP signaling and virulence gene regulation is still unknown.
Surprisingly, we found that deleting all four intact DGCs from the S. flexneri genome reduced c-di-GMP levels but did not eliminate c-di-GMP and that expression of S. flexneri degenerate DGCs dgcE, dgcQ, and dgcN increased c-di-GMP (Fig. 6). The degenerate DGCs in S. flexneri are among those that synthesize c-di-GMP in other Escherichia/Shigella spp (21, 59). Expression of these S. flexneri degenerate DGC genes results in multiple protein isoforms, some of which could potentially contain the GGDEF domain (Fig. 6B). E. coli dgcE is a large protein, encoding ten transmembrane domains within the first 300 residues. It also encodes a predicted MASE1 domain and three PAS domains before a GGDEF and degenerate EAL domain. E. coli DgcE undergoes rapid proteolysis through an unknown protease, resulting in protein degradation. However, this pattern was not observed in the ΔMASE (N terminal) DgcE, suggestive of proteolysis initiating from the N-terminal domain to the C-terminal domain (34, 51). DgcE initiates the expression of biofilm regulator CsgD and mediates curli and cellulose production in E. coli, facilitating exposure to host immune system (60, 61, 62, 63). S. flexneri dgcE contains IS110 family transposase insertion, resulting in a premature stop codon and a frame shift; nevertheless, we found that deletion of dgcE from the Δ4DGC background significantly reduced c-di-GMP. dgcE has been annotated as a pseudogene in nine other pathogenic E. coli strains, including EHEC, EPEC, and EAEC isolates, suggesting a broader evolutionary adaptation associated with pathogenic host colonization (64).
In the case of dgcQ and dgcN, expression of these genes corresponded with two proteins each, one matching the size of the N-terminal reading frame and the other the full-length protein if it had no stop codon. It is unclear how S. flexneri is producing a full-length protein when both of these genes have nonsense mutations. One possibility is translational readthrough (65, 66, 67, 68, 69), or alternatively the bacteria could be inserting selenocysteine into these proteins in lieu of the stop codon. dgcQ mutations have been observed in at least six different strains of EAEC in the same specific pattern observed in S. flexneri 2457T, with an insertion of stop codon at 312 amino acid position and adjacent methionine (AUG) at 314 codon; this suggests that, like dgcE, the dgcQ adaptation is not unique to this specific Shigella strain (64).
S. flexneri dgcM expression in E. coli reduced c-di-GMP levels and was also expressed in two sizes, but these sizes corresponded to the N-terminal and C-terminal portions being separately translated. If one of these two proteins is the DgcM C-terminal portion, it is not clear how they are expressed without a ribosomal binding site. While dgcM does not contain a canonical Shine Dalgarno motif prior to the second start codon, it does contain an A-rich sequence upstream of the second reading frame (AAATAAAT), which can be sufficient to initiate translation (70). Alternatively, dgcM could be translating the second reading frame through translational coupling, where the ribosome translates an adjacent gene, modulated by the gene upstream to it (71, 72).
The mutations in S. flexneri dgcQ, dgcE, and other degenerate DGCs exhibit a very similar pattern, suggesting that this type of mutation is a wider evolutionary mechanism to alter the activity of this enzyme. Deletion of dgcE and dgcQ from the S. flexneri Δ4DGC strain still did not eliminate c-di-GMP in the cell, indicating that other DGCs are still synthesizing the second messenger. It is possible that dgcN or dgcM are capable of c-di-GMP synthesis when expressed in S. flexneri, or the pdeR gene (containing both an EAL and GGDEF domain) has a premature nonsense mutation preceding a GGDEF domain, similar to dgcE and dgcQ.
Experimental procedures
Strain construction and plasmids
Shigella strains used in this study were routinely cultured on tryptic soy broth agar with Congo red, and red colonies were selected to maintain the virulence plasmid (73). Antibiotic concentrations used were as follows until unless otherwise specified: 25 mg/ml ampicillin, 50 mg/ml chloramphenicol, 20 mg/ml gentamicin, 50 mg/ml kanamycin, and IPTG was added at 100 μM. Other details of strains used are summarized in Table S1. S. flexneri Δ4DGC, Δ5DGC, and Δ6DGC mutant strains were generated using homologous recombination (74). Briefly, single genes were deleted by insertion of the cam gene flanked by FRT sites, which were later removed by expressing flippase. DGC expression plasmids were constructed by restriction cloning, using the IPTG inducible pEVS143 (75) as the parental plasmid. The GG→AA mutants were generated by site-directed mutagenesis, using Phusion polymerase (NEB). All strains and plasmids were verified using sequencing. All primers used for homologous recombination and plasmid cloning are listed in Table S2. For infection assays, we used human Henle-407 cells (ATCC CCL-6, HeLa contamination line) as previously described (39).
C-di-GMP quantification using LC-MS/MS method
Overnight cultures from single colonies were grown in LB media with appropriate antibiotics at 30 °C with shaking. Next day, the overnight cultures were subcultured 1:100 in freshly prepared M63 media with Kan and IPTG. The absorbance (A600) of the subcultures grown at 37 °C for ∼3 h was recorded. From this point, the cultures were centrifuged at 4 °C for 5 min. The supernatant was removed and the pellet was resuspended in 500 μl ice cold extraction buffer (40% acetonitrile, 40% methanol, and 0.1 N formic acid). The resuspended pellet was incubated at −20 °C for 30 min and then centrifuged for 20 min at 4 °C. After centrifugation, the liquid was collected and stored at −80 °C until analysis. Prior to analysis, samples were dried using a vacuum concentrator and then rehydrated in 100 μl water. Quantification of c-di-GMP was performed using a Quattro Premier XE mass spectrometer (Waters) coupled with an Acquity high performance liquid chromatography system (Waters) at the Western Michigan University Homer Stryker M.D. School of Medicine (WMeD), at mass 690.69→344.3. Using microscopy, we estimated the mean volume (2.36 × 10^−15^ L) of S. flexneri cells, which was used to determine the intracellular c-di-GMP. The intracellular c-di-GMP concentrations were determined by dividing the c-di-GMP extracted for each strain by the estimated mean volume. Total cell volume of extracted bacteria was calculated by multiplying the CFU/ml to the volume of one bacterium. Experiments were performed in three individual replicates. Graphs were plotted using GraphPad prism (https://www.graphpad.com).
Microscopy for c-di-GMP quantification using riboswitch reporter
S. flexneri strains containing DGC expression plasmids and the biosensor plasmid were grown in LB media with Kan and Amp at 30 °C for ∼16 h. Next day, the strains were subcultured in M63 media with Kan and Amp and were grown for ∼3.5 h. After incubation, the cultures were diluted 1:1000 in M63 media with Kan, Amp, and IPTG. Immediately, 100 μl of this suspension was transferred to a glass-bottom 96-well plate, centrifuged for 10 min at 1000g, and cells were imaged using a BioTek Lionheart FX automated fluorescence microscope for 10 h, with half hour interval. Samples were incubated at 37 °C during imaging. Time zero indicates the first microscopy read after adding IPTG and 10 min centrifugation. Our positive control, the VCA0956 expression strain, was used to optimize the tetramethylrhodamine channel exposure. We used autofocus with a 25 μM Z stack projection and beacons were defined for each well. Fluorescence quantitation was performed using BioTek Gen5 software (https://www.agilent.com/), using fluorescence autothreshold and size threshold between 2 μM and 20 μM. An average cell count for each strain was ∼400 cells at time 0 and ∼4000 cells at the 10th hour for each strain. Experimental quantitation of fluorescence was performed three independent times.
Host cell c-di-GMP kinetics read with microscopy
Henle-407 cells were split in a glass-bottom 96-well plate 2 days before experiment to gain ∼80% confluency (1:3 dilution). One day before experiment, S. flexneri cultures were grown with Kan and Amp in LB media at 30 °C for ∼16 h. On the day of experiment, all the strains were subcultured in M63 media with Kan, Amp, and DOC (0.04% v/v) at 37 °C for ∼4 h. The A650 was measured, and the cells were normalized to 2 × 10^9^ CFU/ml before infection. Infection was performed as previously described (39). Briefly, the normalized cells were used to infect the Henle-407 cells and centrifuged at 105g for 10 min. After centrifugation, the 96-well plate was incubated at 37 °C with 5% CO_2_ for 30 min. After 30 min, the nonadherent population was removed by washing four times with PBS. After washing, 100 μl fresh tissue culture media (FluoroBrite Dulbecco's Modified Eagle's Medium; Thermo Fisher Scientific A1896701) with Kan, Amp, Gen, IPTG, 10 mM Hepes, and 1:1000 dilution Hoechst was added to each well and was immediately imaged for 10 h with half an hour interval. Samples were incubated at 37 °C during imaging. Our positive control VCA0956 was used to setup the exposure settings for Hoechst (blue channel), the tetramethylrhodamine (red channel), and the phase contrast. Settings for analysis and calculations were same as detailed above.
Acid resistance (AR1 and AR2)
AR was setup as previously described with modifications (41, 44). S. flexneri strains were grown overnight in LB media with Kan at 30 °C for ∼16 h. Next day, for AR1 system the strains were subcultured 1:100 in LB media with 50 mM MES, Kan, and IPTG at pH 5.5 overnight at 37 °C with shaking. For AR2, strains were subcultured in LB media with 50 mM MES, 0.4% glucose, Kan, and IPTG at pH 5.5 overnight at 37 °C with no shaking. The following day, the A650 was measured for all the strains (AR1 and AR2) and the bacterial cells were normalized to 5 × 10^8^ CFU/ml. The normalized cells were acid shocked in M9 media at pH 2.5 at 37 °C (AR1) and M9 media at pH 2.5 with 50 mM glutamate at 37 °C (AR2). As a control, each strain was also incubated in M9 media at pH 7. After 1 h, cells were plated with serial dilution. Colony counts and the dilution factor was recorded to calculate the percent survival, determined by dividing the CFU/ml of acid shocked strains by the CFU/ml of the same strains incubated in pH seven medium.
Cell culture assays
The invasion assay was set up as previously described (39). Briefly, 1 day before experiment, Henle-407 were split in 6-well plates for ∼10% confluency. S. flexneri DGC strains were inoculated in LB media with Kan and incubated overnight at 30 °C. Each strain was subcultured 1:100 in LB media with Kan, IPTG, and 0.04% DOC for ∼4 h. The A600 was normalized to 2 × 10^9^ CFU/ml and 100 μl of suspension was added to individual wells to infect Henle cells. Plates were centrifuged for 10 min at 1000 rpm and then incubated for 30 min at 37 °C with 5% CO_2_. Each well was washed four times with PBS and incubated again with Gentamycin for 45 min. After washing the cells twice with PBS, each well was stained with Giemsa stain for 5 min and washed with water to remove excess stain. Later, 300 cells were counted for positively invaded cells with three or more bacterial cells.
Plaque assay was setup using previously described protocols (39). Briefly, 2 days before experiment, Henle-407 cells were split in 6-wells plates for ∼80% confluency. Overnight grown strains were subcultured 1:100 in LB with Kan for ∼4 h. The cells were normalized to 5 × 10^4^ CFU/ml, and 100 μl was added to individual wells. Each 6-well plate was centrifuged and incubated for 45 min at 37 °C with 5% CO_2_. Each well was washed four times with PBS and incubated with Kan, IPTG, and 20% glucose for 48 h. After incubation, wells were washed twice with PBS and stained with crystal violet for 2 min. All plates were later imaged and plaque size was measured using ImageJ software (https://imagej.net/ij/).
Immunoblotting
Immunoblots were performed as previously described (20). Bacterial cultures were grown in LB broth at 37 °C. When the cells reached the mid log phase of growth, the A600 was read, and cells were collected by centrifugation and resuspended in SDS sample buffer (5% β-mercaptoethanol, 3% (wt/vol) SDS, 10% glycerol, 0.02% bromophenol blue, 63 mM Tris-Cl, pH 6.8) at a concentration of 2 × 10^9^ CFU/ml or 4 × 10^9^ CFU/ml (for dgcN and dgcE only). Samples were boiled for 10 min and then electrophoresed in a 20% SDS-PAGE gel. After electrophoresis, proteins were transferred to a 0.45-μm pore size nitrocellulose membrane (Hybond-ECL; GE Healthcare) and incubated with a α-HIS antibody (Genscript A00186), followed by horseradish peroxidase-conjugated α-mouse antibody (Abclonal AS003). Signals were detected by developing the blot with a Pierce ECL detection kit (Thermo Fisher Scientific). Protein domain predictions were made using MistDB (76).
Data availability
We confirm that the data supporting the findings of this study are available within the article and its supplementary materials, and raw data are available from the corresponding author (B. K.) upon request.
Supporting information
This article contains supporting information (38).
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Kotloff K.L.Winickoff J.P.Ivanoff B.Clemens J.D.Swerdlow D.L.Sansonetti P.J.Global burden of Shigella infections: implications for vaccine development and implementation of control strategies Bull. World Health Organ.77199965166610516787 PMC 2557719 · pubmed ↗
- 2Bennish M.L.Wojtyniak B.J.Mortality due to Shigellosis: community and hospital data Clin. Infect. Dis.131991 S 245S 25110.1093/clinids/13.supplement_4.s 2452047645 · doi ↗ · pubmed ↗
- 3Mantis N.Prévost M.C.Sansonetti P.Analysis of epithelial cell stress response during infection by Shigella flexneri Infect. Immun.64199624742482869846910.1128/iai.64.7.2474-2482.1996 PMC 174100 · doi ↗ · pubmed ↗
- 4Maurelli A.T.Blackmon B.Curtiss R.Temperature-dependent expression of virulence genes in Shigella species Infect. Immun.431984195201636089510.1128/iai.43.1.195-201.1984 PMC 263409 · doi ↗ · pubmed ↗
- 5CDC Antibiotic Resistance Threats in the United States 2013 U.S. Department of Health and Human Services Atlanta, GA
- 6CDC Increase in Extensively Drug-Resistant Shigellosis in the United States 2023 U.S. Department of Health and Human Services Atlanta, GA
- 7Jennison A.V.Verma N.K.Shigella flexneri infection: pathogenesis and vaccine development FEMS Microbiol. Rev.28200443581497552910.1016/j.femsre.2003.07.002 · doi ↗ · pubmed ↗
- 8Dorman C.J.The virulence plasmids of Shigella flexneri Schwartz E.Microbial Megaplasmids 2009 Springer Berlin Heidelberg Berlin, Heidelberg 151170