RET is a sex-biased regulator of intestinal tumorigenesis
Sean T. Koester, Naisi Li, Neelendu Dey

TL;DR
The RET gene influences intestinal tumor growth differently in males and females, with the effect mediated by gut microbes.
Contribution
RET is identified as a sex-biased regulator of tumorigenesis influenced by the microbiome in an Apc-deficient CRC model.
Findings
ApcMin/+Ret+/− females had significantly more tumors than males, a sex bias not seen in ApcMin/+ mice.
Antibiotics reduced tumor burden and reversed the sex bias, while microbiome reconstitution restored it.
RET and TLR4 expression were correlated in female but not male tumor samples.
Abstract
Ret is implicated in colorectal cancer (CRC) as both a proto-oncogene and a tumor suppressor. We asked whether RET signaling regulates tumorigenesis in an Apc-deficient preclinical model of CRC. We observed a sex-biased phenotype: ApcMin/+Ret+/− females had significantly greater tumor burden than ApcMin/+Ret+/− males, a phenomenon not seen in ApcMin/+ mice, which had equal distributions by sex. Dysfunctional RET signaling was associated with gene expression changes in diverse tumor signaling pathways in tumors and normal-appearing colon. Sex-biased gene expression differences mirroring tumor phenotypes were seen in 26 genes, including the Apc tumor suppressor gene. Ret and Tlr4 expression were significantly correlated in tumor samples from female but not male ApcMin/+Ret+/− mice. Antibiotics resulted in reduction of tumor burden, inverting the sex-biased phenotype such that…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
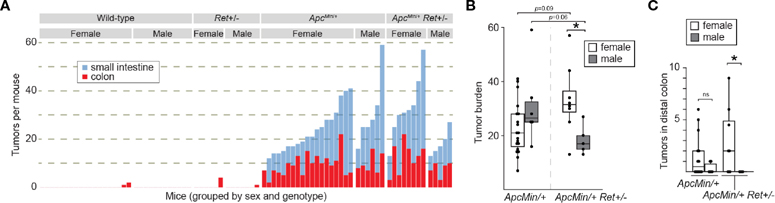 Figure 1
Figure 1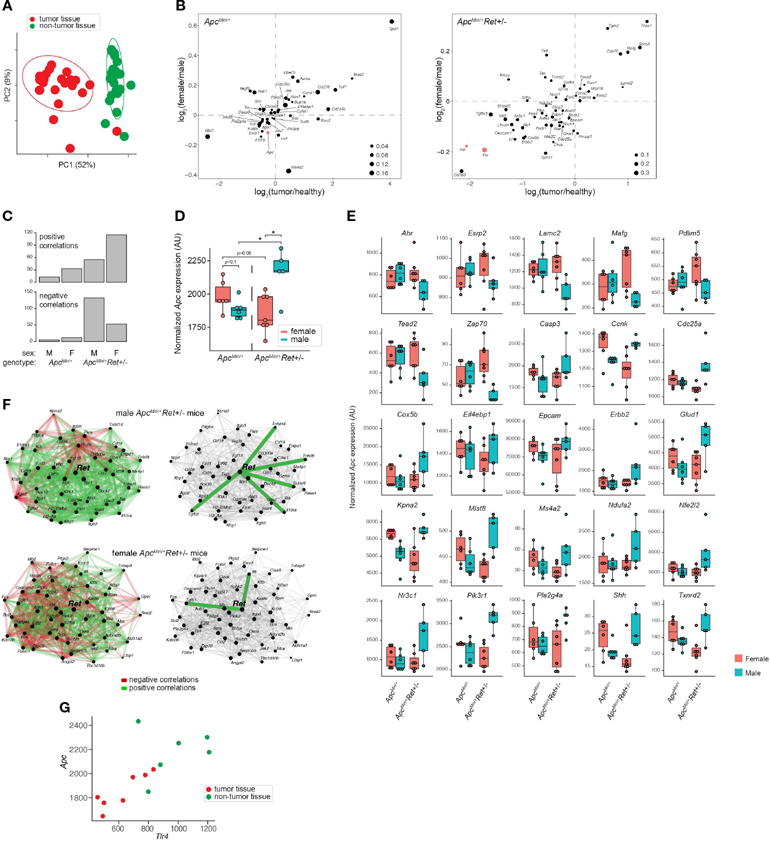 Figure 2
Figure 2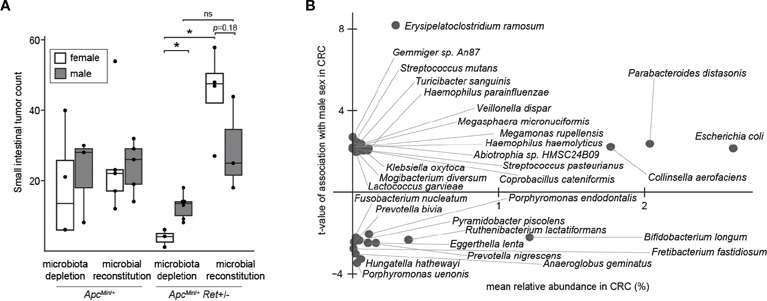 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic factors in colorectal cancer · Cancer Immunotherapy and Biomarkers · Heat shock proteins research
Introduction
Colorectal cancer (CRC) is one of the most common cancers globally. Ret, which is critical in enteric nervous system (ENS) development and maintenance, has been implicated as both a proto-oncogene (1) and tumor suppressor (2) in CRC. While RET fusions in metastatic CRC portend a poor prognosis (3), specific RET inhibitors such as selpercatinib and pralsetinib have demonstrated efficacy in targeting oncogenic RET rearrangements [reviewed at (4, 5)]. Apc encodes a tumor suppressor and is commonly mutated in CRC. We assessed interactions of Apc and Ret through a crossbreeding experiment using Apc^Min/+^ mice and Ret+/− mice (6). We administered 1.5% dextran sodium sulfate (DSS) to induce colonic tumors in Apc^Min/+^Ret+/− progeny (as well as littermate controls with a mutation in Apc only, Ret only, or neither). We expected that Apc^Min/+^ mice would develop many intestinal tumors compared to wild-type or Ret+/− mice, which are not predisposed to developing tumors, but it was unknown whether mice harboring mutations both in Apc and Ret would develop a greater, lesser, or equivalent number of tumors compared to Apc^Min/+^ mice. We hypothesized that if tumorigenesis is regulated by RET signaling, then we should observe a change in tumor burden in Apc^Min/+^Ret+/− mice that is either decreased compared with Apc^Min/+^ mice if RET signaling promoted tumorigenesis or increased if RET suppressed tumorigenesis.
Results
Consistent with expectations, (i) Apc^Min/+^ mice (n=25) had a significantly greater tumor burden than wild-type littermates (n=31) (mean ± SEM: 22.6 ± 2.2 vs 0.16 ± 0.12, p<10^−8^ [females]; 31.2 ± 6.1 vs 0 ± 0, p=0.004 [males]; Figures 1A, B; Supplementary Table 1); (ii) Ret+/− mice (n=13) had tumor burdens not significantly different from wildtype littermates (n=31) (0.7 ± 0.7 vs 0.2 ± 0.1 [females]; 0.1 ± 0.1 vs 0 ± 0, [males]; Figure 1A; Supplementary Table 1); and (iii) compared to Apc^Min/+^ mice that did not receive DSS (n=30), DSS-treated Apc^Min/+^ mice developed more colonic tumors (9.3 ± 1.0 vs 1.1 ± 0.3, p<10^−6^ [females]; 10 ± 1.7 vs 1.8 ± 0.3, p=0.004 [males]). In the experimental Apc^Min/+^Ret+/− cohorts, there was no overall significant difference in tumor burden compared to Apc^Min/+^ mice (two-way ANOVA; Figure 1A). However, we observed a sex-biased interaction of Ret and Apc: total (colonic plus small intestinal) tumor burden was significantly greater in female Apc^Min/+^Ret+/− mice compared to male Apc^Min/+^Ret+/− mice (3-way ANOVA, interaction between Apc, Ret, and sex covariates: p<0.003, F_1,74 =_ 10; Figure 1B). Compared to males, female Apc^Min/+^Ret+/− mice had significantly greater tumor burden in the distal colon, which we defined as the last 25% of the colon by length (2.94 ± 1.17 vs 0 ± 0, p<0.04, Student’s two-tailed t-test; Figure 1C). In the small intestine, Apc^Min/+^Ret+/− females had more tumors than males (20.5 ± 4.2 vs 10.6 ± 2.3, p=0.06, Student’s two-tailed t-test). These sex-biased differences did not exist among Apc^Min/+^ mice.
Male Apc^Min/+^Ret+/− mice had longer small intestines than female Apc^Min/+^Ret+/− mice (36.4 ± 0.37 cm vs 33.9 ± 0.85 cm; p<0.03, Student’s two-tailed t-test). While of unclear biological significance, it excludes small intestinal length as a potential confounder for the difference in small intestinal tumor burden, which was greater in females. Otherwise, there were no sex-based differences in tumor size, intestinal lengths, or whole gut transit times (Supplementary Table 1). Within control cohorts, there were also no sex-based differences in these metrics.
To define Ret’s role in tumor signaling pathways, we profiled gene expression in colonic tumors (n=24) and healthy-appearing colonic tissues (n=24) from male and female Apc^Min/+^ and Apc^Min/+^Ret+/− mice (Supplementary Table 2A). Global gene expression profiles clustered predominantly by whether they represented tumors or healthy tissues (p=0.001, PERMANOVA [permutational multivariate analysis of variance using distance matrices] using Bray-Curtis dissimilarity; Figure 2A). Expression levels of a subset of genes were nonetheless affected by the interaction between sex, genotype, and tissue sample type (ANOVA: p<0.05; Figure 2B). Consistent with their established biological roles, Apc, Fxr (Wnt pathway antagonist; bile acid receptor), and Vdr (vitamin D receptor; bile acid receptor) were expressed at higher levels in healthy tissues. In tumors, Myc (oncogene), Lgr5 (stem cell marker), and Wnt5a (Wnt pathway activator) were expressed at higher levels and were affected by the interaction between sex and genotype (ANOVA: p<0.05). Consistent with Fxr’s role as a negative regulator of bile acid synthesis, total bile acid concentrations were higher in females than males (3,604.2 ± 418.2 ng/mg stool vs 2,410.84 ± 177.2 ng/mg stool, p=0.01, Student’s two-tailed t-test).
Ret deficiency appeared to dysregulate homeostatic gene expression networks, resulting in a dramatic increase in numbers of genes significantly correlated with Apc expression in tumor samples (Figure 2C). Interestingly, Apc^Min/+^Ret+/− female mice had more positive than negative correlations, while Apc^Min/+^Ret+/− male mice had more negative than positive correlations (Figure 2C). The genes correlated with Apc are implicated in diverse pathways, suggesting global effects on gene expression (Supplementary Table 2B). In healthy samples, Apc expression was greater in Apc^Min/+^Ret+/− males than in females (2,452 ± 58 arbitrary units [AU] vs 2,181 ± 83 AU, p<0.03, Student’s two-tailed t-test). In tumor samples, Apc^Min/+^Ret+/− males had significantly higher Apc expression than both Apc^Min/+^Ret+/− females (2,160 ± 80 AU vs 1,854 ± 55 AU, p<0.02, Student’s two-tailed t-test) and Apc^Min/+^ males (2,160 ± 80 AU vs 1,885 ± 29 AU, p=0.02, Student’s two-tailed t-test). Notably, within tumor samples, Apc expression was higher in male Apc^Min/+^Ret+/− mice than in female Apc^Min/+^Ret+/− mice, and this trend was opposite in Apc^Min/+^ mice — the inverse of the sex-biased genotype-dependent tumor phenotype (Figure 2D). Including Apc, tumor-intrinsic expression levels of 26 genes mirrored tumor phenotypes (exactly or inversely) and were significantly affected by interactions between sex and genotype (p<0.05, two-way ANOVA) (Figure 2E). While Apc expression levels varied with tumor phenotypes in such a manner that it may plausibly explain the underlying biology, the biological significance of the other 25 genes is more challenging to interpret and underscores complexity of gene network interactions.
Strikingly, tumor-intrinsic Ret-centric gene expression networks were entirely non-overlapping between male and female Apc^Min/+^Ret+/mice (Figure 2F). In tumors harvested from Apc^Min/+^Ret+/− males, we observed correlations between Ret and Il10ra, Tnfsf14, Tnfsf8, Tnfrsf4, and Tgfb2 (r≥0.89, p<0.05, Pearson correlation), which are involved in anti-inflammatory responses. In contrast, in Apc^Min/+^Ret+/− females, Ret was significantly correlated with Tgfb1 and with Il6 (r≥0.78, p<0.05, Pearson correlation), both of which are involved in upregulating the immune system. These findings suggest links between the immune system and Ret, whereby loss of functional RET signaling leads to a pro-inflammatory state in females and an anti-inflammatory state in males.
Intriguingly, Ret expression was significantly correlated with Tlr4 expression in tumor samples from all cohorts of mice (r≥0.79, p<0.04, Pearson correlation) except for the male Apc^Min/+^Ret+/− mice. TLR4 is critical in microbial pattern recognition and is overexpressed in colorectal cancers and adenomas compared to healthy tissues in humans (7). Tlr4 was correlated with Apc in Apc^Min/+^Ret+/− female mice (tumor samples: r=0.88, p<0.01; all samples: r=0.73, p<0.005, Pearson correlation; Figure 2G) but not in other cohorts.
Thus, we next tested the role of the gut microbiome in driving sex bias in tumor phenotypes. We either administered chronic antibiotics or first depleted the native microbiota with antibiotics and then reconstituted the microbiota via oral gavage using a uniform fecal suspension. Chronic antibiotics reduced tumor burden, significantly more so in females, and resulted in complete inversion of the sex-biased tumor phenotype: microbiota-depleted Apc^Min/+^Ret+/− females had significantly fewer tumors than males (3.7 ± 1.5 vs 12.7 ± 1.5, p=0.005, two-tailed Student’s t-test; Figure 3A). The interaction between sex and microbiome status (harboring or lacking a microbiome) was significant (p<0.02, F1,12 = 7, two-way ANOVA).
Finally, we asked whether human CRC data support our findings. Approximately 10% of CRCs are reported as harboring Ret mutations (2). Intriguingly, in The Cancer Genome Atlas (8), CRCs with Ret mutations occurred predominantly in females (z=2.98, p<0.003). Further, in a reanalysis of published CRC microbiome surveys (9), we found that 68 bacterial species were differentially abundant between sexes in CRC (Figure 3B); of these, only 11 species were also differentially abundant in the healthy cohort, evidencing CRC-specific sex biases. E. coli was more abundant in males with CRC than in females with CRC, whereas in the healthy cohort, E. coli was more abundant in females than in males. In contrast, Fusobacterium nucleatum was more abundant in females than in males in the CRC cohort. F. nucleatum, which is linked to CRC progression and metastasis, has been associated with CpG methylation and a CpG island methylator phenotype, which is associated with the female sex (10).
Discussion
The answer to our initial question — is RET an oncogene (1) or tumor suppressor (2) in CRC? — appears to be complicated: it depends on sex and the microbiome. Underlying the phenotype that we discovered were significant differences in tumor burden in the distal colon. Interestingly, public datasets suggest that Ret mutations in CRC and CRC microbiome signatures are both sex-biased. Our study offers proof-of-principle that CRC risk-modulating gut microbial effects depend on sex and genetics, and they underscore the importance of evaluating sex as a biological variable in research and of reporting the sexes of both human and non-human study participants.
A limitation of this study is that the specific cellular players remain unknown. Our gene expression data suggest that both tumor-intrinsic and tumor-extrinsic cells are disrupted by Ret insufficiency, and that the resulting Ret gene networks starkly differ by sex. RET is a critical player in the ENS, which transmits intestinal growth signals via the GLP-2 pathway (11, 12), potentially facilitates CRC metastasis (13, 14), and regulates immunity (15) and physiology (16). Enteric neurons express estrogen receptor (17) so could form the basis for sex-biased microbiome-dependent tumorigenesis. Future studies should elucidate tumor cell-autonomous effects versus effects of a RET-deficient ENS. This research could lead to novel personalized CRC prevention tactics.
Materials and methods
Animal husbandry
Male and female Apc^Min/+^, Ret+/−, and Apc^Min/+^Ret+/− mice (C57BL/6 background) were studied using methods approved by the Institutional Animal Care and Use Committee of Fred Hutchinson Cancer Center (protocol 51049). Mice from the same litter were co-housed regardless of differences in genotype, thereby leading to shared microbiota due to coprophagy. Mice were fed ad libitum with PicoLab Mouse Diet 5058. DSS (1.5% by volume) was administered in drinking water for 7 days to mice at 6–8 weeks of age. Following DSS exposure, mice were given standard drinking water for the remainder of the experiment. Mice were euthanized ~6 weeks after DSS exposure (or earlier if exhibiting symptoms of high tumor burden) using aerosolized 1.5% isoflurane delivered through a precision vaporizer. At euthanasia, small intestines and colons were harvested, rinsed in ice cold phosphate buffered saline to remove fecal material, and opened longitudinally for tumor counting and characterization. Fresh fecal pellets were snapfrozen in liquid nitrogen and stored at −80°C until use. Tumor localization analysis. Digital images of colons were equally split into quartiles. Tumors spanning different segments of the colon had fractions of tumors counted in each segment. These fractions were estimated as 0.25, 0.5, or 0.75, and the fractions for each tumor add up to 1. Microbiome manipulation studies. Vancomycin (1 g/L), metronidazole (1 g/L), and neomycin (0.5 g/L) antibiotics (Millipore Sigma, St. Louis, MO) were delivered via drinking water containing 2% sucrose (20 g/L) over 10 days or, in the experiment involving chronic antibiotic administration, cyclically throughout the duration of the experiment. Recolonization was performed via oral gavage of a fecal suspension from female Apc^Min/+^Ret+/− mice. Due to enhanced DSS toxicity in the setting of antibiotic use, mice receiving antibiotics were not given DSS; therefore, small intestinal tumor counts were used as readouts in these experimental cohorts.
Gut transit time measurements
Using previously published methods (18), mice were gavages with 200 μL per mouse of a sterilized 6% red carmine dye solution (Sigma C1022) and monitored for time to initial passage per rectum. Gavages were performed by the same individual (N.L.) within a consistent time frame in all mice in order to minimize variability.
RNA isolation
After flushing with PBS, gut segments were stored in RNAlater (Thermo Fisher Scientific Inc., Waltham, MA) at 4°C for 24 hours and then transferred to −20°C for storage until use. To isolate RNA, 20 mg of each sample was placed into a tube containing 0.1 mm zirconium beads, a 4 mm steel ball, and homogenization buffer before mechanical disruption using a TissueLyser II (Qiagen, Hilden, Germany). RNA was purified using RNeasy Mini kits (Qiagen).
Colonic tumor and non-tumor gene expression profiling
Gene expression data were generated using NanoString nCounter^®^ Tumor Signaling 360 Panels in conjunction with a 55-gene Panel Plus.
Analysis of published human microbiome datasets
We utilized curatedMetagenomicData, an R package linked to a database of curated data from nearly 100 microbiome surveys (9). We filtered in stool samples from individuals with CRC and healthy individuals at least 18 years of age, none of whom reported current antibiotic use. We identified 625 stool samples from individuals with CRC (392 males and 233 females) and 5,221 stool samples from otherwise healthy individuals (2,178 males and 3,043 females). To control for differences in age and study population, we included only the subset of individuals from the healthy cohort who were (i) from the same countries represented in the CRC cohort and (ii) within 2 years of age of an individual in the CRC cohort. Ultimately, this resulted in a healthy cohort comprised of 1,707 individuals (911 males and 796 females). We then used corncob, an R package that uses beta-binomial regression to model relative abundances and identify bacteria significantly associated with covariates of interest (19), to identify bacterial species that were significantly differentially abundant in CRC between sexes.
Data analysis
Statistical comparisons were performed in R (version 4.0.0). Figures were generated using R using native functions as well as the ggplot2 (version 3.1.0) and pheatmap (version 1.0.12) packages. Figures were assembled in Adobe Illustrator.
Supplementary Material
Supplementary Table 1 | Individual mouse data. Unique mouse identification numbers, cage identification numbers, sex, genotype, age at time of euthanasia, microbiota, whether DSS was used, diet, transit times, small intestinal length, cecal length, colonic length, and tumor counts by location.SUPPLEMENTARY TABLE 1 Individual mouse data. Unique mouse identification numbers, cage identification numbers, sex, genotype, age at time of euthanasia, microbiota, whether DSS was used, diet, transit times, small intestinal length, cecal length, colonic length, and tumor counts by location.
Supplementary Table 2 | Gene expression data. (A) Normalized data from the NanoString nCounter® Tumor Signaling 360 Panels in conjunction with a 55-gene Panel Plus. (B) Genes significantly correlated with Apc expression in ApcMin/+ and ApcMin/+Ret+/- mice.SUPPLEMENTARY TABLE 2 Gene expression data. (A) Normalized data from the NanoString nCounter^®^ Tumor Signaling 360 Panels in conjunction with a 55-gene Panel Plus. (B) Genes significantly correlated with Apc expression in Apc^Min/+^ and Apc^Min/+^Ret+/− mice.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Guy R Association between germline mutations in the RET proto-oncogene and colorectal cancer and polyps. Adv Res Gastroenterol Hepatol (2019) 12:44–47. doi: 10.19080/ARGH.2019.12.555835 · doi ↗
- 2Luo Y, Tsuchiya KD, Il Park D, Fausel R, Kanngurn S, Welcsh P, RET is a potential tumor suppressor gene in colorectal cancer. Oncogene (2013) 32:2037–47. doi: 10.1038/onc.2012.22522751117 PMC 3465636 · doi ↗ · pubmed ↗
- 3Pietrantonio F, Di Nicolantonio F, Schrock AB, Lee J, Morano F, FucàG, RET fusions in a small subset of advanced colorectal cancers at risk of being neglected. Ann Oncol (2018) 29:1394–401. doi: 10.1093/annonc/mdy 09029538669 · doi ↗ · pubmed ↗
- 4Takahashi M RET receptor signaling: Function in development, metabolic disease, and cancer. Proc Jpn Acad Ser B (2022) 98:112–25. doi: 10.2183/pjab.98.00835283407 PMC 8948417 · doi ↗ · pubmed ↗
- 5Desilets A, Repetto M, Yang S-R, Sherman EJ, Drilon A. RET-altered cancers—A tumor-agnostic review of biology, diagnosis and targeted therapy activity. Cancers (2023) 15:4146. doi: 10.3390/cancers 1516414637627175 PMC 10452615 · doi ↗ · pubmed ↗
- 6Enomoto H, Crawford PA, Gorodinsky A, Heuckeroth RO, Johnson EM, Milbrandt J. RET signaling is essential for migration, axonal growth and axon guidance of developing sympathetic neurons. Development (2001) 128:3963–74. doi: 10.1242/dev.128.20.396311641220 · doi ↗ · pubmed ↗
- 7Sewda K, Coppola D, Enkemann S, Yue B, Kim J, Lopez AS, Cell-surface markers for colon adenoma and adenocarcinoma. Oncotarget (2016) 7:17773–89. doi: 10.18632/oncotarget.740226894861 PMC 4951249 · doi ↗ · pubmed ↗
- 8The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature (2012) 487:330–7. doi: 10.1038/nature 1125222810696 PMC 3401966 · doi ↗ · pubmed ↗