GRIEVOUS: your command-line general for resolving cross-dataset genotype inconsistencies
James V Talwar, Adam Klie, Meghana S Pagadala, Hannah Carter

TL;DR
GRIEVOUS is a command-line tool that simplifies aligning genetic data across datasets to ensure consistency in studies like genome-wide association analyses.
Contribution
GRIEVOUS introduces a novel command-line tool for resolving genotype inconsistencies across datasets using an internal database and custom indexing.
Findings
GRIEVOUS identifies and aligns biallelic SNPs across summary statistic and genotype files.
The tool extracts the maximal set of biallelic SNPs common to all datasets after harmonization.
GRIEVOUS is freely available and can be installed via pip for easy use.
Abstract
Harmonizing variant indexing and allele assignments across datasets is crucial for data integrity in cross-dataset studies such as multi-cohort genome-wide association studies, meta-analyses, and the development, validation, and application of polygenic risk scores. Ensuring this indexing and allele consistency is a laborious, time-consuming, and error-prone process requiring a certain degree of computational proficiency. Here, we introduce GRIEVOUS, a command-line tool for cross-dataset variant homogenization. By means of an internal database and a custom indexing methodology, GRIEVOUS identifies, formats, and aligns all biallelic single nucleotide polymorphisms (SNPs) across all summary statistic and genotype files of interest. Upon completion of dataset harmonization, GRIEVOUS can also be used to extract the maximal set of biallelic SNPs common to all datasets. GRIEVOUS and all…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
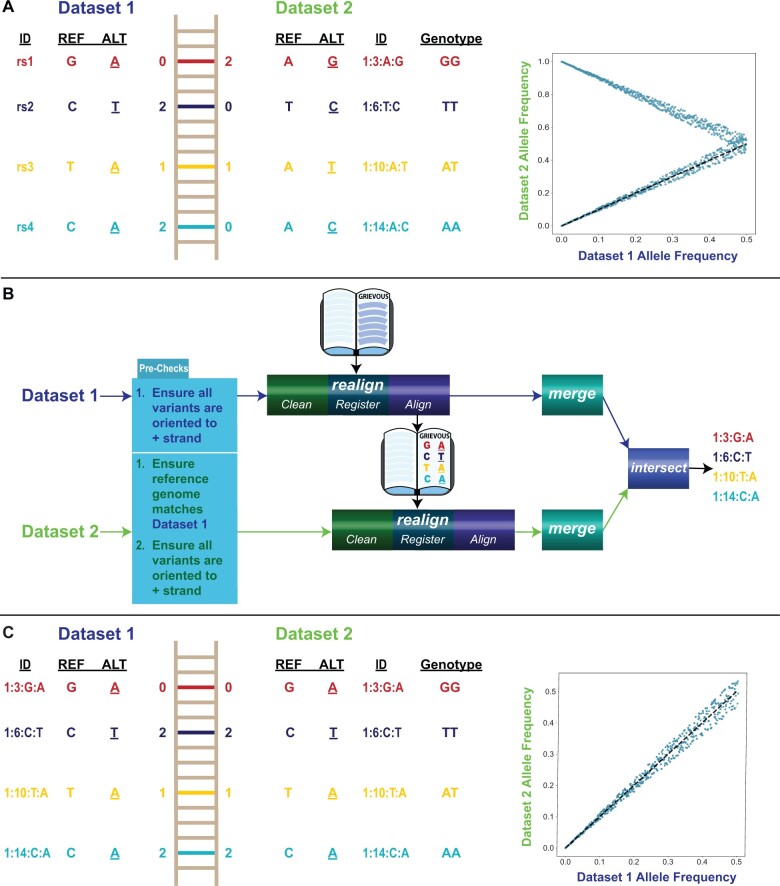 Figure 1
Figure 1| Condition | Dataset | Total number of variants | Number of biallelic SNPs identified | Number of biallelic SNPs reassigned | Average run time (minutes) | Max memory usage (GB) | Cross-dataset SNP intersection size |
|---|---|---|---|---|---|---|---|
| Breast cancer | Michailidou | 11 792 542 | 10 413 027 | 0 | 1.71 ± 1.08 | 3.00 | 10 295 993 |
| DRIVE | 48 838 144 | 44 849 194 | 2 | 9.13 ± 4.03 | 9.97 | ||
| UKBB | 97 013 422 | 92 429 914 | 659 | 8.03 ± 3.69 | 14.77 | ||
| Prostate cancer | Conti | 29 235 255 | 26 529 293 | 0 | 4.66 ± 3.48 | 6.80 | 26 098 297 |
| ELLIPSE | 48 899 508 | 44 905 050 | 11 329 170 | 7.22 ± 3.49 | 8.55 | ||
| UKBB | 97 013 422 | 92 424 521 | 11 374 523 | 10.42 ± 7.85 | 14.96 |
- —National Institutes of Health10.13039/100000002
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic Associations and Epidemiology · Genomics and Rare Diseases · Bioinformatics and Genomic Networks
1 Introduction
The explosion of genetic variant datasets, driven by increasingly affordable genomic profiling technologies, presents exciting opportunities across a number of fields, ranging from precision medicine to ecology to agriculture. However, taking full advantage of genetic data from multiple sources can be challenging when datasets use different conventions for encoding variants. Analyses requiring the integration of various datasets range from the straightforward, such as validating genetic findings or polygenic scores in new datasets, to the more complex, such as cross-dataset joint statistical analyses [e.g. genome wide association meta-analyses (Zeggini and Ioannidis 2009, Evangelou and Ioannidis 2013), two-sample Mendelian randomizations (Hartwig et al. 2016, Sanderson et al. 2022)]. These approaches can frequently be undermined by discrepancies in the definition of identical variants across datasets (Zeggini and Ioannidis 2009, Hartwig et al. 2016).
Common variants, such as single nucleotide polymorphisms (SNPs), are abundantly shared across different datasets, and can be imputed against reference panels to increase coverage (van Leeuwen et al. 2015, Coleman et al. 2016). Quality control of SNP data is critical, and best practices include setting uncertain genotypes to missing, orienting genotypes to the forward reference DNA strand, and filtering on missingness, minor allele frequency, or deviation from Hardy Weinberg equilibrium (Anderson et al. 2010, Coleman et al. 2016, Marees et al. 2018, Choi et al. 2020). However, even after performing the steps to generate a high-quality dataset, there can still be discrepancies between datasets due to differences in conventions used to define genotypes. If left uncorrected, these discrepancies can create illusory genotypic differences where none exist. Given that common variant sets can include millions of SNPs, it can be challenging and time consuming to manually identify and resolve discrepancies.
To reduce this burden to the researcher, we developed a Generalized Realignment of Innocuous and Essential Variants Otherwise Utilized as Skewed or GRIEVOUS, a command-line tool designed to ensure cross-cohort consistency and maximal feature recovery of biallelic SNPs, the most commonly used class of variants for genetic studies (Choi et al.2020). Whether creating a composite cohort from smaller studies for joint analyses, or ensuring feature fidelity for validation studies or polygenic score portability, GRIEVOUS reduces the problem of variant consistency and recovery to a simple streamlined set of commands.
2 Materials and methods
2.1 Datasets
Summary statistics and genotype data were obtained for two diseases: breast cancer (BC) and prostate cancer (PC). Specifically, data consisted of one summary statistic file and two different genotype datasets, each of which diverged in genotyping and imputation methodology from the other. Each file (i.e. both summary statistics and genotypes), for each condition studied here, indexed variants in a unique manner, capturing the discordance that can exist between datasets.
For BC, summary statistics were obtained from a large-scale BC GWAS study by Michailidou et al. (2017) (downloaded from: https://bcac.ccge.medschl.cam.ac.uk/bcacdata/oncoarray/oncoarray-and-combined-summary-result/gwas-summary-results-breast-cancer-risk-2017), and the genotype datasets utilized were from the Discovery, Biology, and Risk of Inherited Variants in Breast Cancer (DRIVE; dbGaP study accession: phs001265.v1.p1) project and the UK Biobank (UKBB). For PC, summary statistics from a large-scale PC multi-ancestry GWAS meta-analysis by Conti et al. (2021) were downloaded from dbGaP (study accession: phs001120.v2.p1). Genomic data from the ELucidating Loci Involved in Prostate cancer SuscEptibility (ELLIPSE; dbGaP study accession: phs001120.v1.p1) consortium and UKBB were utilized as the PC genotype datasets. PC PRSs were calculated as an effect size weighted genotype summation, with effect sizes and P-values for SNP subselection derived from the Conti et al. (2021) summary statistics. Finally, we note that both DRIVE and ELLIPSE were subsets of each disease’s summary statistic GWAS.
Both DRIVE and ELLIPSE were genotyped using the OncoArray microarray (Amos et al. 2017), while the UKBB was genotyped using the UK Biobank Axiom Array (Bycroft et al. 2018). To recover untyped markers in both DRIVE and ELLIPSE, we imputed genotypes with Minimac4 using the 1000 Genomes Phase 3 Version 5 reference panel, via the Michigan Imputation Server (Das et al.2016). Details of UKBB imputation can be found in the original UKBB report by Bycroft et al. (2018)
2.2 GRIEVOUS: design and framework
The current state of variant indexing can be summarized concisely as unsystematic. Datasets can use any means or mechanisms for indexing variants, ranging from custom concatenations of dataset variant information (e.g. CHR_POS_REF_ALT), to rsIDs, to any mixture in between (Fig. 1A). Arbitrary assignments of reference (REF) and alternate (ALT) definitions across datasets poses a similar problem (Fig. 1A). Differing genotyping technologies, and in the case of summary statistics, the characterization of effect alleles, often lead to divergences in REF/ALT definitions across datasets. Harmonization of dataset indices and allele assignments, thus first requires variant organization, a task specifically addressed by the design of GRIEVOUS (Fig. 1B).
(A) Example of the same genotypes described by two different genomic datasets that use different schemas for indexing and assigning variants. This complicates variant recovery and leads to spurious artifactual allele frequency divergences across datasets. (B) Each genomic dataset is harmonized with GRIEVOUS. Upon ensuring all datasets use the same reference assembly and strand orientation, each dataset is passed through realign, sequentially, and then passed to merge to generate composite dataset level reports of all identified biallelic and inverted variants resulting from the GRIEVOUS realignment process. Finally, intersect is called once across all datasets, to identify the maximal set of biallelic SNPs common across all datasets. (C) After harmonization with GRIEVOUS, all genomic datasets use the same schema of variant indexing, and all biallelic SNPs common across all datasets are consistently assigned, eliminating cross-dataset artifactual allele frequency divergences.
GRIEVOUS deploys a flexible internal database, backed by chromosome-level parquet files, to build a unified variant index and align allele assignments. Initialized in an empty manner, GRIEVOUS databases are continuously updated with each run, adding all hitherto unobserved biallelic SNPs identified in a given dataset (Fig. 1B). The iterative nature of database variant additions ensures that each subsequent dataset is consistently oriented with the set of common biallelic SNPs found in all previously GRIEVOUS realigned datasets.
The key command behind GRIEVOUS is realign, which consists of three steps: clean, register, and align (Supplementary Fig. S1). Clean identifies valid biallelic SNPs, by means of a graph formulation (Supplementary Methods: Identifying Valid Variants), and resolves duplications and multiple indexing issues. Specifically, variant positions are defined as nodes and dataset-specific IDs as edges. Any node for which there exists an edge to another node in the graph is considered an invalid variant, with a dataset-specific ID pointing to multiple genomic positions. Register performs variant indexing reformulation (i.e. a colon-separated unification of chromosome, position, REF, and ALT defined by the dataset; CHR:POS:REF:ALT) and database comparison. Dataset variants found in the reverse index assignment from a GRIEVOUS database are marked for realignment, while all variants not found in the database by genomic position are registered for addition. Align enacts the adjustment of SNP assignments, updates the database, and writes output files for the realign run. These files include reports of identified, reassigned, and duplicated variants, and for PLINK2 binary file inputs(Purcell et al. 2007, Chang et al.2015), reassignment files to convert the binary genomic data to the GRIEVOUS format.
GRIEVOUS can harmonize an arbitrary number of user-defined genomic datasets, whether they be PLINK2 binary files, summary statistics, or a mixture of the two (Fig. 1C). Variants are processed at the chromosome-level, allowing parallelization and integration into different workflows. Additional commands, merge and intersect, combine chromosome-level reports and generate the maximal set of biallelic SNPs common to all datasets of interest, respectively (Fig. 1B). GRIEVOUS also supports the creation of multiple databases for project-specific needs, enhancing parallel processing and organization.
3 Results
Polygenic risk scores (PRS) provide an example of an application where performance and portability depend on the fidelity of SNP assignments across datasets. They are traditionally calculated as an effect size weighted genotype summation, wherein each SNP’s effect size is multiplied by the number of corresponding alleles carried, and these products are summed across risk alleles. Thus, if differences exist in how genotypes are defined across datasets, the contribution of discrepant SNPs to the score could be calculated incorrectly, introducing noise to the PRS calculation and reducing score performance on the outcome of interest.
This problem can be avoided through careful harmonization of SNP consistency across datasets prior to PRS calculation. GRIEVOUS accomplishes this through indexing unification, assignment synchronization, and variant recovery across datasets. To demonstrate GRIEVOUS’ effectiveness, we applied it to harmonize summary statistics and genotype data for two different diseases: breast cancer (BC) and prostate cancer (PC; Materials and methods: Datasets).
For each condition, GRIEVOUS realign was applied sequentially in a dataset consistent order: (i) summary statistics, (ii) DRIVE/ELLIPSE genotypes, (iii) UKBB genotypes. This process aligned the ALT allele in the genotyped datasets to the effect allele in the summary statistics for all biallelic variants, facilitating unbiased cross-disease comparison of SNP reassignments. The results are presented in Table 1.
Both ELLIPSE and DRIVE used different nomenclature for common variant indexing relative to the UKBB, a disparity that GRIEVOUS effectively managed. Post-reindexing, GRIEVOUS successfully recovered and reassigned all database-consistent biallelic SNPs across datasets. For BC, there was high concordance in variant assignment, likely because DRIVE was part of the BC GWAS. However, in PC, despite ELLIPSE being part of the PC GWAS, 43.4% of cross-dataset common variants differed in allele assignment from the summary statistics and required reassignment. These discrepancies drastically impacted PRS generalization in both PC genotype datasets. Specifically, PRSs applied to the unharmonized PC datasets exhibited an area under the receiver operating characteristic curve (AUC) below random chance. However, after harmonizing all datasets with GRIEVOUS, expected PRS performance was restored (Supplementary Table S1).
In summary, GRIEVOUS harmonized datasets and identified the shared set of biallelic SNPs for both diseases, demonstrating its utility in genomic dataset harmonization.
4 Conclusion
The harmonization of variant indexing and assignments is crucial for the accuracy of multi-dataset studies of genetic variants such as the development, validation, and application of PRSs. Here we introduce GRIEVOUS, a command-line tool designed to simplify and expedite the process of harmonizing and recovering variants across different cohorts. GRIEVOUS efficiently unifies variant indices, and primarily focuses on the recovery of biallelic SNPs. Variants not fitting this category currently still require manual verification by the user to ensure allele assignment consistency. However, these could be supported in future releases. Before using GRIEVOUS, it is important for users to confirm that all datasets use the same reference assembly and strand orientation to ensure accurate results.
In summary, GRIEVOUS streamlines the time-consuming and meticulous process of SNP homogenization and reduces opportunities for human error.
Supplementary Material
btae489_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Amos CI , Dennis J, Wang Z et al The Onco Array Consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol Biomarkers Prev 2017;26:126–35.27697780 10.1158/1055-9965.EPI-16-0106 PMC 5224974 · doi ↗ · pubmed ↗
- 2Anderson CA , Pettersson FH, Clarke GM et al Data quality control in genetic case-control association studies. Nat Protoc 2010;5:1564–73.21085122 10.1038/nprot.2010.116PMC 3025522 · doi ↗ · pubmed ↗
- 3Bycroft C , Freeman C, Petkova D et al The UK Biobank resource with deep phenotyping and genomic data. Nature 2018;562:203–9.30305743 10.1038/s 41586-018-0579-z PMC 6786975 · doi ↗ · pubmed ↗
- 4Chang CC , Chow CC, Tellier LC et al Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015;4:7.25722852 10.1186/s 13742-015-0047-8PMC 4342193 · doi ↗ · pubmed ↗
- 5Choi SW , Mak TS-H, O'Reilly PF et al Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc 2020;15:2759–72.32709988 10.1038/s 41596-020-0353-1PMC 7612115 · doi ↗ · pubmed ↗
- 6Coleman JRI , Euesden J, Patel H et al Quality control, imputation and analysis of genome-wide genotyping data from the Illumina Human Core Exome microarray. Brief Funct Genomics 2016;15:298–304.26443613 10.1093/bfgp/elv 037PMC 5863770 · doi ↗ · pubmed ↗
- 7Conti DV , Darst BF, Moss LC et al Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat Genet 2021;53:65–75.33398198 10.1038/s 41588-020-00748-0PMC 8148035 · doi ↗ · pubmed ↗
- 8Das S , Forer L, Schönherr S et al Next-generation genotype imputation service and methods. Nat Genet 2016;48:1284–7.27571263 10.1038/ng.3656 PMC 5157836 · doi ↗ · pubmed ↗