DNA Methylation in Noncancerous Liver Tissues as Biomarker for Multicentric Occurrence of Hepatitis C Virus–Related Hepatocellular Carcinoma
Hiroyuki Suzuki, Hideki Iwamoto, Ken Yamamoto, Mai Tsukaguchi, Toru Nakamura, Atsutaka Masuda, Takahiko Sakaue, Toshimitsu Tanaka, Takashi Niizeki, Shusuke Okamura, Shigeo Shimose, Tomotake Shirono, Yu Noda, Naoki Kamachi, Ryoko Kuromatsu, Toru Hisaka, Hirohisa Yano

TL;DR
This study identifies DNA methylation changes in noncancerous liver tissues that may predict the multicentric recurrence of HCV-related liver cancer after surgery.
Contribution
The study reveals 9 genes with altered DNA methylation in noncancerous tissues linked to multicentric recurrence of HCC.
Findings
Nine gene regions showed significant DNA methylation differences between early and nonearly recurrence groups.
These methylation changes were consistent across two independent patient sets.
The findings suggest a potential biomarker for predicting multicentric HCC recurrence.
Abstract
Hepatitis C virus (HCV)-related hepatocellular carcinoma (HCC) progresses with a highly multicentric occurrence (MO) even after radical hepatectomy. Despite several efforts to clarify the pathogenesis of MO, the underlying molecular mechanism remains elusive. The aim of this study was to evaluate alterations in DNA methylation in noncancerous liver tissues in the MO of HCC. A total of 203 patients with HCV-related HCC who underwent radical hepatectomy at our hospital between January 2008 and January 2012 were recruited. We defined a group of nonearly recurrence of HCC (NR) for ≥3 years after radical hepatectomy and a group of early recurrence of HCC (ER) with MO within 2 years after radical hepatectomy. Three patients each were selected in the NR and ER groups in the first set, and 13 patients in the NR group and 17 patients in the ER group were selected in the second set. Genome-wide…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1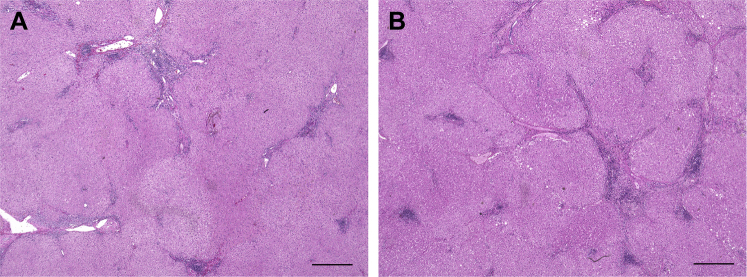 Figure 2
Figure 2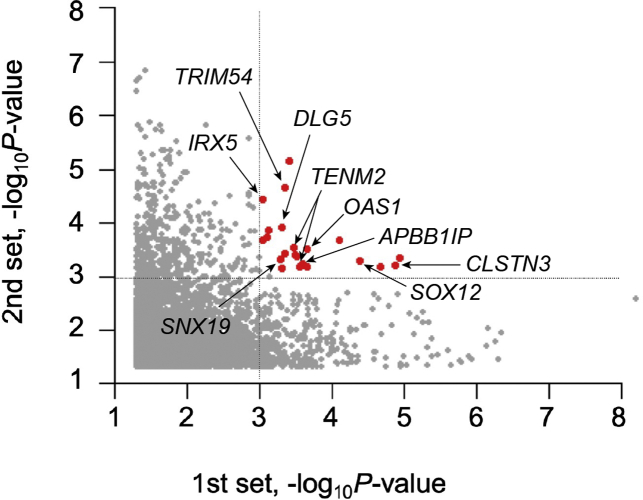 Figure 3
Figure 3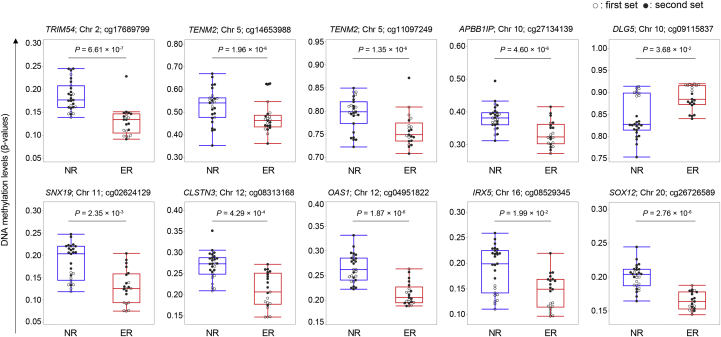 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLiver Disease Diagnosis and Treatment · Hepatitis C virus research · Epigenetics and DNA Methylation
Introduction
Hepatocellular carcinoma (HCC) is the most common primary liver cancer and the third leading cause of cancer-related deaths worldwide.1^,^2 HCC typically occurs in the setting of persistent hepatitis or cirrhosis secondary to hepatitis B virus and hepatitis C virus (HCV) infection, alcoholism, or nonalcoholic steatohepatitis.2 Epidemiologic evidence has indicated that persistent infection with HCV is a major risk factor for developing HCC, and cirrhosis is the main cause of HCV-related HCC; the core protein of HCV has also been considered to be directly involved in hepatocarcinogenesis.3 The high recurrence rate of HCV-related HCC is dependent on its characteristics, such as intrahepatic metastasis and multicentric occurrence (MO).1 Even after radical treatment, such as hepatectomy and radiofrequency ablation, the low disease-free survival rates of HCV-related HCC (5 years, 24%) result in high mortality rate and limited prognosis.4^,^5 Therefore, identifying patients at high risk of HCC recurrence after radical treatment is important. One of the mechanisms that cause MO in HCV-related HCC, even after sustained virologic response (SVR), is the epigenetic alteration of hepatocytes in the liver caused by chronic hepatitis.6^,^7
In the last few decades, epigenetic alterations have been shown to be almost universal among different types of cancers and have become increasingly important for understanding human malignancies.8 DNA methylation, a major epigenetic alteration, regulates the gene transcription and stability without affecting the DNA sequence.8 Aberrant accumulation of DNA methylation, caused by a long-term exposure to carcinogens and chronic inflammation, is known to form a carcinogenic field in a variety of malignancies and to play an important role in carcinogenesis; for example, methylation within the promoter region of tumor-suppressor genes causes their silencing and methylation within the gene itself may trigger mutational events.9 Genome-wide DNA methylation profiling of various types of cancers, including HCC, has shown that substantial alteration in DNA methylation plays an important role in the setting of carcinogenesis.10 Aberrant methylation of promoter CpG islands in several genes has been reported to be a possible biomarker for HCV-related HCC development; however, the detailed mechanisms of the relationship between MO and DNA methylation have not been clarified.11, 12, 13 Furthermore, there are no comprehensive reports comparing the histologic DNA methylation levels in noncancerous liver tissues between cases with and without early MO after radical hepatectomy.
The aim of this study was to verify whether alterations in histologic DNA methylation in noncancerous liver tissues of HCV-related HCC are involved in the MO.
Methods
Patients
Tissue samples from 203 patients with HCV-related HCC were obtained by radical hepatectomy at our hospital between January 2008 and January 2012. The exclusion criteria were as follows: (1) HCC recurrence pattern exhibiting characteristics of intrahepatic metastasis (recurrent within 3 months after radical hepatectomy); (2) under systemic steroid therapy, which is known to directly affect DNA methylation14^,^15; (3) inadequate follow-up after radical hepatectomy; and (4) HCC previously treated with systemic chemotherapy. Eventually, a total of 36 patients were selected and were assigned to 2 groups: nonearly recurrence group (NR), patients without HCC recurrence, >3 years after hepatectomy; and early recurrence group (ER), patients with multicentric recurrence of HCC within 3 years after hepatectomy. We performed analysis of the 2 sets; in the first set experiments, 3 cases each from the NR and ER groups were selected, and 3 different tissues were collected from each patient. At least 9 tissues from each group were evaluated. In the second set of experiments, 13 and 17 cases from the NR and ER groups, respectively, were selected. In the second set, one tissue sample was collected from each patient (Figure 1). Noncancerous liver tissues were taken from the resected tissues when performed radical hepatectomy. These tissues were immediately frozen in liquid nitrogen and kept at −80 °C until the following DNA extracting preparation.Figure 1. Flowchart of this study.
DNA Methylation Chip Assay
After extracting DNA from noncancerous liver tissues, DNA methylation levels of about 470,000 CpG methylation sites located on the autosome were determined using an Illumina Human Methylation 450 BeadChip (Illumina, San Diego, CA) according to the manufacturer’s instructions. For 401,282 CpG methylation sites, excluding CpG sites related to single nucleotide polymorphisms or sites with poor data acquisition rate, the differences in DNA methylation levels between the NR and ER groups were analyzed using the generalized linear model without any adjustments.16 Gene expression levels in the liver were obtained from the Genotype-Tissue Expression project.17
Statistical Analysis
Data are expressed as numbers or medians. Differences between the 2 groups were analyzed using the Wilcoxon signed-rank test. P values <.05 and <.001 were considered to indicate statistically significant and extremely significant differences, respectively. The difference in DNA methylation levels was tested using a generalized linear model, and significant differences between the ER and NR groups were determined after Bonferroni correction. Data analyses were performed using R Statistical Software (version 2. 15. 2; R Foundation for Statistical Computing, Vienna, Austria).
Ethics Statement
This study was performed in accordance with the principles of the Declaration of Helsinki and was approved by the ethical committee of the Kurume University School of Medicine (Ethical code: 144) and the Institutional Review Board of Kyushu University (Ethical code: 538-00).
Results
Patient Characteristics
In the first set, the median age was 67.9 years, and 66.7% of the patients were male (Table 1). In the second set, the median age was 72.2 years, and 63.3% of the patients were male (Table 2). There were no significant differences with respect to the presence of diabetes, Child-Pugh score, prevalence of SVR, tumor characteristics (such as serum tumor markers, tumor number, and tumor differentiation), and histopathological findings between the NR and ER groups (Tables 1 and 2); whereas, in the second set, the patients in the ER group were younger than those in the NR group (68.4 vs 75.2; P = .032; Table 2). In the first set, the median recurrence-free observation period was 1390 (range, 1285–1421) days for the NR group and 419 (range, 370–469) days for the ER group (Table 1). The median recurrence-free observation period was 1449 (range, 1111–2474) days for the NR group and 784 (range, 270–914) days for the ER group in the second set (Table 2). The representative histologic image of the cases from ER and NR groups is shown in Figure 2, whose degree of HCV-related cirrhosis was F2A2.Table 1. Baseline Clinical and Tumor Characteristics for the First SetClinical characteristicsTotal (n = 6)NR (n = 3)ER (n = 3)P valueAge (y)67.963.171.6.067Gender, male/female4/22/12/11.000Diabetes mellitus +/−4/21/23/0.083Child-Pugh score 5/64/22/12/11.000Liver damage A/B4/22/12/11.000AST (IU/L)73.542.7104.3.140ALT (IU/L)33.732.035.3.819Total bilirubin (mg/dL)0.740.660.79.56Platelet (×10^3^/μL)12.712.912.4.865SVR +/−2/40/32/1.051Tumor characteristics TNM classification0/4/22/1/02/1/01.000 Number 1/2/34/1/11/1/13/0/0.148 Tumor size (mm)25.229.321.7.200 Alpha-fetoprotein (ng/mL)462.830.0903.3.371 DCP (mAU/mL)1140.82258.323.3.372Pathological findings Fibrosis 1/2/3/41/0/2/31/0/0/20/0/2/1.101 Activity 1/20/60/30/31.000 Differentiation well/moderate/poor2/4/01/2/01/2/01.000 Macroscopic classification SN/SNEG/CMN/others3/0/1/21/0/1/12/0/0/1.422Clinical time course Median recurrence-free observation period (d)15991390419P values were obtained by comparing between the NR and ER groups.AFP, alpha-fetoprotein; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CMN, confluent multinodular type; DCP, des-γ-carboxy prothrombin; SN, simple nodular type; SNEG, simple nodular type with extranodular growth.Table 2. Baseline Clinical and Tumor Characteristics for the Second SetClinical characteristicsTotal (n = 30)NR (n = 13)ER (n = 17)P valueAge (y)72.268.475.2.032Gender male/female19/116/713/4.088Diabetes mellitus +/−19/116/713/4.088Child-Pugh score 5/620/107/612/5.346Liver damage A/B18/129/49/8.301AST (IU/L)51.151.850.6.940ALT (IU/L)53.151.754.0.888Total bilirubin (mg/dL)0.760.820.71.360Platelet (×10^3^/μL)14.715.414.1.500SVR +/−4/263/101/16.170Tumor characteristics TNM classification5/20/52/11/03/9/5.084 Number 1/2/323/6/112/1/011/5/1.199 Tumor size (mm)28.730.327.8.650 Alpha-fetoprotein (ng/mL)167.0166.6172.8.973 DCP (mAU/mL)1140.82258.323.3.512Pathological findings Fibrosis 1/2/3/47/6/6/114/5/1/33/1/5/8.051 Activity 1/26/243/103/14.713 Differentiation well/moderate/poor3/23/42/11/01/12/4.299 Macroscopic classification SN/SNEG/CMN/others21/5/1/39/2/0/212/3/1/1.693Clinical time course Median recurrence-free observation period (d)11161449784P values were obtained by comparing between the NR and ER groups.ALT, alanine aminotransferase; AST, aspartate aminotransferase; CMN, confluent multinodular type; DCP, des-γ-carboxy prothrombin; SN, simple nodular type; SNEG, simple nodular type with extranodular growth.Figure 2. Representative histopathological findings. Representative pathologic images of the noncancerous liver tissues from (A) NR and (B) ER groups (hematoxylin and eosin staining). Scale bars represent 500 μm.
DNA Methylation Profiling Across the Genome
Among the ER and NR groups, 4798 and 5651 CpG methylation sites were significantly different in the first and second sets, respectively. Among these CpG methylation sites, 22 were found to have significant changes in common between the 2 sets (Figure 3). Excluding genes with different directional methylation (hypo vs hyper) between the first and second sets, 10 CpG methylation sites and 9 genes were identified for common directional methylation changes between the 2 sets (Table 3). The methylation level of each gene is shown in Figure 4. In the methylation analysis, we observed a potential functional role of hypomethylation in 8 HCV-related HCC that affected their genes (TRIM54, TENM2, APBB1IP, SNX19, CLSTN3, OAS1, IRX5, and SOX12) between the NR and ER groups in common between the 2 sets (Table 3). From the Genotype-Tissue Expression project,15 the highest expression level in the liver was observed for CLSTN3 (25.84 transcripts per million), followed by that for APBB1IP (18.84 transcripts per million) and SNX19 (13.12 transcripts per million; Table 3).Figure 3. Dots plots for differences in CpG methylation sites. Common significant differences between the 2 sets. Red plots represent the 22 CpG sites, which showed commonly significant differences between the 2 groups (P < .001).Table 3. Ten CpG Cites With the Same Direction of DNA Methylation and Significantly Different Between the 2 SetsChromosomePositionCpG siteGeneFirst setSecond setCombineGene expression in the liver (TPM)BetaP valueBetaP valueBetaP value227,529,540cg17689799TRIM54−0.0564.59 × 10^−4^−0.0552.20 × 10^−5^−0.0536.61 × 10^−7^0.0925167,171,284cg14653988TENM2−0.0763.12 × 10^−4^−0.1044.13 × 10^−4^−0.0931.96 × 10^−6^0.3715167,394,019cg11097249TENM2−0.0492.82 × 10^−4^−0.0496.47 × 10^−4^−0.0501.35 × 10^−6^0.3711026,681,347cg27134139APBB1IP−0.0573.30 × 10^−4^−0.0554.01 × 10^−4^−0.0544.60 × 10^−6^18.841079,541,592cg09115837DLG50.0135.08 × 10^−4^0.0431.17 × 10^−4^0.0273.68 × 10^−2^2.23211130,786,230cg02624129SNX19−0.0415.20 × 10^−4^−0.0474.94 × 10^−4^−0.0402.35 × 10^−3^13.12127,315,531cg08313168CLSTN3−0.0651.35 × 10^−5^−0.0396.32 × 10^−4^−0.0454.29 × 10^−4^25.8412113,345,598cg04951822OAS1−0.0452.22 × 10^−4^−0.0483.11 × 10^−4^−0.0461.87 × 10^−6^7.1651654,971,700cg08529345IRX5−0.0249.07 × 10^−4^−0.0423.69 × 10^−5^−0.0311.99 × 10^−2^0.01820306,364cg26726589SOX12−0.0324.15 × 10^−5^−0.0294.99 × 10^−4^−0.0292.76 × 10^−6^3.356TPM, transcripts per million.Figure 4DNA methylation levels at 10 CpG sites. White dots represent patients in the first set, and black dots represent patients in the second set.
Discussion
In the present study, we observed that 9 gene regions, viz., APBB1P, CLSTN3, DLG5, IRX5, OAS1, SOX12, SNX19, TENM2, and TRIM54, exhibited significant differences in the DNA methylation levels (P < .001) in the common direction between the ER and NR groups in the 2 analysis sets.
Eight of these 9 genes had higher DNA methylation levels in the ER group compared with that in the NR group. In general, it is known that the higher the number of methylation sites contributes worse prognosis in patients with cancer. Our results are consistent with those presented in a recent report on “proliferation class” of HCC, which is characterized as a clinically aggressive tumor.18 We evaluated the correlation between MO and DNA methylation by examining the function of genes whose DNA methylation levels were lower in the ER group compared with those in the NR group.
TRIM54, a member of the tripartite motif family, is thought to be involved in several biological processes, such as carcinogenesis, cellular growth, and cellular differentiation.19 Recently, using RNA-sequencing data of 292 HCC patients from The Cancer Genome Atlas, TRIM54 was reported to be one of the vascular invasion-associated 14-gene signature to predict the prognosis in patients with HCC.20 In addition, TRIM54 has been reported to affect the expression of p62/IMP2, which is an essential regulator of Wnt signaling pathways and plays an important role in the progression and metastasis of HCC.21
In patients with malignant ovarian cancer, decreased TENM2 expression is significantly associated with shortened overall survival.22 It has been reported that compared with primary cultured human hepatic stellate cells, alcohol-injured human hepatic stellate cells exhibit downregulation of the expression of TENM2.23 As these conditions can precede HCC, the presence of early genetic alterations might be considered as an initiating event implicated in carcinogenesis. In line with these findings, we show that hypomethylation of TENM2 is associated with the early recurrence of HCC.
ABPP1IP, identified as a Rap1-binding protein that is important in the activation of leukocyte integrin, is known to be involved in the infiltration ability of leukocytes.24 Although there are no reports on the relationship between APBB1IP and HCC or HCV infection, several studies have revealed the relationship between APBB1IP and malignancies. In a study on clear cell renal cell carcinoma, circular-APBB1IP was reported to be a prognostic biomarker promoting tumor growth and invasion through the activation of ERK1/2 signals.25 In another study on colorectal cancer using cell-free DNA, hypermethylation of APBB1IP was identified as a novel candidate for cancer detection.26 Furthermore, in a study on gastric cancer, 19 serum proteins, including APBB1IP, were used to distinguish patients with and without cancer.27
SNX19, a central regulator of cell trafficking and signal transduction, has been identified as one of the genes associated with diabetes and schizophrenia.28 Although there is no report on the relationship between SNX19 and HCV infection or HCC, it has been reported that 18 genes, including SNX19, were significantly overexpressed in thyroid oncocytic adenomas compared with their expression in controls.29
CLSTN3, a member of evolutionarily conserved synaptogenic adhesion proteins of the cadherin superfamily, was identified as a synaptic adhesion molecule that acts through presynaptic neurexins.30CLSTN3 is known to regulate neural regulation of energy and bone homeostasis.31 CLSTN3 was reported to be upregulated in patients with hepatocellular adenoma compared with that in patients with HCC, and CLSTN3 was recognized to be involved with the difference in the degree of HCV-related cirrhosis (in the common term between; F2 vs F4 and F3 vs F4, F1 vs F3).32^,^33
OAS1 is an interferon-stimulating gene that plays an important role in the innate immune response, cell differentiation, and apoptosis.34 Single nucleotide polymorphisms in OAS1 are associated with susceptibility to chronic HCV infection, progression of hepatic fibrosis, hepatic necroinflammatory activity grade, and development of HCC.35^,^36 The expression of OAS1 was negatively correlated with breast and prostate cancer progression, whereas it was positively correlated with prognosis in patients with colorectal cancer.37^,^38 Because OAS1 is strongly associated with HCV infection and tumor prognosis, it is reasonable that there is a correlation between changes in methylation in noncancerous liver tissue and MO.
IRX5 plays pivotal roles in normal embryonic cell patterning and in the development of malignancies.39 IRX5 was reported to be upregulated in HCC tissues compared with that in adjacent normal tissues, and it promotes the proliferation of HCC cells by upregulating the expression of cyclin D1.40 Several studies regarding the association of IRX5 with cancers have shown that depending on the type of cancer, IRX5 could act both in tumor-suppressing and tumor-promoting roles via the transforming growth factor beta signaling.41^,^42 In the present study, IRX5 was hypomethylated in the NR group compared with that in the ER group; therefore, further studies are warranted to clarify the function of IRX5 in noncancerous tissues.
SOX12 is a member of the sex-determining region Y–related high mobility group box family of transcription factors that is characterized by the presence of a highly conserved DNA-binding high mobility group domain. In HCC, Sox12 is one of the cancer stem cell markers.43 The expression of SOX12 is correlated with the malignancy of HCC (such as extracapsular invasion, microvascular invasion, and lymph node metastasis), and higher SOX12 levels are associated with poorer prognosis in HCC patients (overall survival P = .0004, progression-free survival P = .0013).44^,^45 Specifically, SOX12 activates the expression of Twist1 and fibroblast growth factor–binding protein 1, which promote the epithelial-mesenchymal transition of HCC and increase the expression of CDK14 and insulin-like growth factor 2 mRNA-binding protein 1, defining the malignant phenotype of HCC.44^,^46 Furthermore, some cancer-related microRNAs (miR), such as miR-874 and miR-744, have been reported to directly target SOX12 and suppress its action.47^,^48 Further elucidation of the relationship with DNA methylation in SOX12 is desired.
In the present study, DLG5 was hypermethylated in the group with MO. It has been reported that DLG5 is downregulated in HCC, and lower DLG5 expression is associated with poor survival of patients with HCC, and that the accumulation or overexpression of DLG5 inhibits the proliferation and intrahepatic and lung metastasis of HCC cells.49^,^50
The present study had several limitations, including its single-center retrospective design and the small sample size. In addition, although DNA methylation is generally considered a stable epigenetic mark, there are possibilities of false positives and ignoring local changes. Furthermore, there were significant differences for DLG5 and IRX2 when the first and second sets were independently assessed (P < .001); however, when the 2 sets were combined, the difference became smaller and exhibited the same tendency. This may be because the methylation levels of these genes were separated in the first and second sets (Table 3). Unlike the fact that a very high SVR rate was achieved using direct-acting antivirals in clinical practice, the SVR rate was only 20% for the patients in this study.51 However, because epigenetic alterations remain present even after SVR obtained by interferon or direct-acting antiviral treatment, it is important to evaluate epigenetic alterations regardless of the achievement of SVR.7^,^52 The elimination of these limitations requires the accumulation of clinical cases, which could enable more accurate prediction of HCV-related HCC recurrence.
In conclusion, after radical hepatectomy of HCV-related HCC, methylation levels at particular CpG sites in DNA from noncancerous liver tissues appear to be suitable biomarkers for predicting MO of HCC. Further validation in a large cohort of patients is required.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sung H.Ferlay J.Siegel R.L.Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries CA Cancer J Clin 7120212092493353833810.3322/caac.21660 · doi ↗ · pubmed ↗
- 2El-Serag H.B.Rudolph K.L.Hepatocellular carcinoma: epidemiology and molecular carcinogenesis Gastroenterology 1322007255725761757022610.1053/j.gastro.2007.04.061 · doi ↗ · pubmed ↗
- 3Koike K.Hepatitis C virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways J Gastroenterol Hepatol 22 Suppl 12007 S 108S 1111756745710.1111/j.1440-1746.2006.04669.x · doi ↗ · pubmed ↗
- 4Sasaki Y.Yamada T.Tanaka H.Risk of recurrence in a long-term follow-up after surgery in 417 patients with hepatitis B- or hepatitis C-related hepatocellular carcinoma Ann Surg 24420067717801706077110.1097/01.sla.0000225126.56483.b 3PMC 1856577 · doi ↗ · pubmed ↗
- 5Takeishi K.Maeda T.Tsujita E.Predictors of intrahepatic multiple recurrences after curative hepatectomy for hepatocellular carcinoma Anticancer Res 3520153061306625964596 · pubmed ↗
- 6Ding X.He M.Chan A.W.H.Genomic and epigenomic features of primary and recurrent hepatocellular carcinomas Gastroenterology 157201916301645.e 63156089310.1053/j.gastro.2019.09.005 · doi ↗ · pubmed ↗
- 7Hamdane N.Jühling F.Crouchet E.HCV-induced epigenetic changes associated with liver cancer risk persist after sustained virologic response Gastroenterology 156201923132329.e 73083609310.1053/j.gastro.2019.02.038PMC 8756817 · doi ↗ · pubmed ↗
- 8Baylin S.B.Jones P.A.A decade of exploring the cancer epigenome - biological and translational implications Nat Rev Cancer 1120117267342194128410.1038/nrc 3130 PMC 3307543 · doi ↗ · pubmed ↗