Genetic aspects of ataxias in a cohort of Turkish patients
Basak Gogus, Muhsin Elmas, Ulku Turk Boru

TL;DR
This study examines the genetic causes of ataxia in Turkish patients, highlighting the role of whole exome sequencing in identifying underlying mutations.
Contribution
The study provides insights into the genetic etiology of ataxia in a Turkish cohort, emphasizing the prevalence of autosomal recessive and dominant inheritance patterns.
Findings
Autosomal recessive inheritance was detected in 80.5% of patients, while autosomal dominant inheritance was found in 19.5%.
Five patients had unique genetic variants in genes such as MCM3AP, AGTPBP1, GDAP2, and SH3TC2.
Abnormal cerebellum and polyneuropathy were observed in MRI and EMG tests, respectively.
Abstract
Ataxia is one of the clinical findings of the movement disorder disease group. Although there are many underlying etiological reasons, genetic etiology has an increasing significance thanks to the recently developing technology. The aim of this study is to present the variants detected in WES analysis excluding non-genetic causes, in patients with ataxia. Thirty-six patients who were referred to us with findings of ataxia and diagnosed through WES or other molecular genetic analysis methods were included in our study. At the same time, information such as the onset time of the complaints, consanguinity status between parents, and the presence of relatives with similar symptoms were evaluated. If available, the patient’s biochemical and radiological test results were presented. Thirty-six patients were diagnosed through WES or CES. The rate of detected autosomal recessive inheritance…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
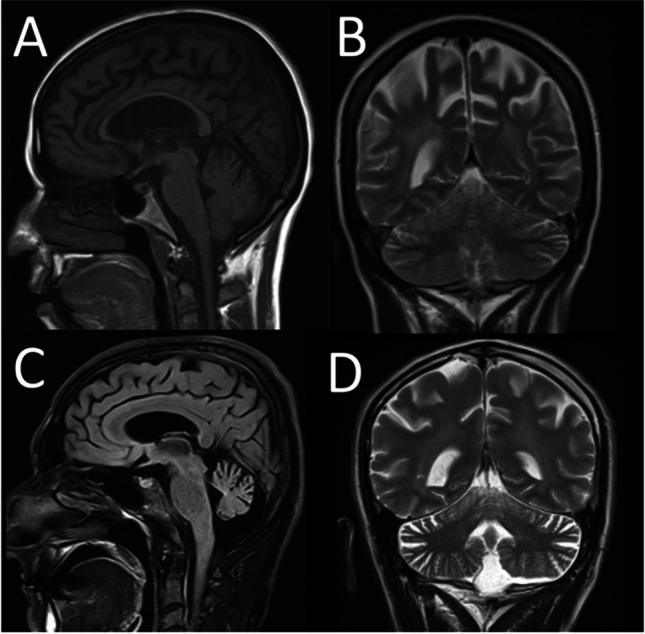 Figure 1
Figure 1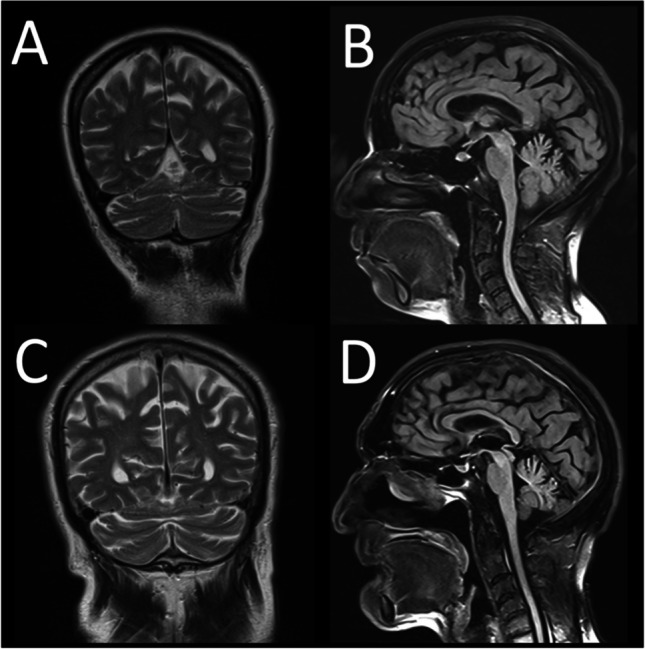 Figure 2
Figure 2- —Afyonkarahisar Health Science University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic Neurodegenerative Diseases · Neurological diseases and metabolism · Hereditary Neurological Disorders
Introduction
Ataxia originated from the Greek word, meaning “disorderly’ or “lack of order.” It is a general term formerly used for heartbeat, gait, and movement disorders. The definition of ataxia is loss of function such as gait abnormality, speech changes, and abnormalities in eye movements [1]. The causes for this damage are ischemic or hemorrhagic cerebellar strokes, alcohol, toxins, medications, infections, multiple sclerosis, thyroid problems, autoimmune diseases, head trauma, paraneoplastic cerebellar degeneration, brain tumors, or genetic mutation.
Clinical exome sequencing (CES) and whole exome sequencing (WES) analyses are frequently performed while diagnosing genetic diseases, most of which are neurological diseases [2]. Whole exome sequencing (WES) is a genomic technique for sequencing all protein-coding regions of genes in a genome (known as the exome). Humans have approximately 180,000 exons, constituting nearly 1% of the human genome, or approximately 30 million base pairs. The human exome contains ~ 85% of known disease-related variants. The remaining 15% of disease-causing variants are located within introns (non-coding regions) [3]. CES, on the other hand, consists of targeted next-generation sequencing (NGS) panels containing approximately 4800–5000 genes associated with known clinical phenotypes.
In OMIM (Online Mendelian Inheritance in Man), which is one of the largest databases of genetic diseases, there are 926 phenotypes that include “ataxia” and have known molecular basis. Genetic approach, especially next-generation sequencing analysis, has therefore become important in ataxia patients. Studies have shown that 52–35% of ataxia patients have been diagnosed through new-generation sequencing analysis [4–7].
The aim of this study is to present the variants detected in WES analysis performed in patients with ataxia. Non-genetic causes were excluded in these patients. This study supports the importance of genetic evaluation in ataxia patients when all etiologic causes are excluded. At the same time, the study presents diversity of degree and variability on phenotype of complex diseases accompanied with ataxia.
Methods
The aim of this study is to present clinical and genetic data of ataxia patients diagnosed through next-generation sequencing analysis. This is a retrospective study and single-center experience. For this purpose, the patient file was reviewed and re-evaluated. The patient criteria to be included in this study are as follows:
- Referral to Afyonkarahisar Health Sciences University (AFSU), Department of Medical Genetics from the Afyonkarahisar Health Sciences University (AFSU), Department of Neurology between 2012 and 2023
- To have genetic results over 18 years old, acquired through only next-generation sequencing analysis like WES
- To have signs of ataxia in physical examination
- Not have acquired ataxias and nongenetic ataxias such as those related to infection, trauma, or stroke
The exclusion criteria are as follows:
- Patients who did not have WES or CES results as well as other genetic tests were not included in the study
- Patients whose WES or CES result did not reveal a variant that would explain the patient’s symptoms were not included in the study
Thirty-six patients were included in the study, according to these criteria. The anamnesis information of the patients was evaluated via the patient database information system of AFSU. The onset time of the complaints of the patients, presence of consanguinity between the parents, and similar cases in the family were questioned. Detailed physical examination findings of the patients were evaluated. Biochemical and radiological (magnetic resonance imaging, computed tomography, etc.) test results of the patients were reviewed using the patient database information system. In addition, electromyography and additional diagnostic test results of the patients were examined, if available.
WES analyses of the patients were performed by contracted institutions. The variants responsible for the phenotype of the patients were reported to us. Segregation analysis was not performed on the patients. The classification of variants by the American College of Medical Genetics was evaluated with Franklin database. Changes detected with pathogenic, likely pathogenic and variant of significance categories, were considered. If mutations in the gene with detected variant caused more than one disease in the OMIM database, differential diagnosis was performed with physical examination and test results. Possible genetic etiology responsible for ataxia signs was determined for the patients.
Following the exclusion of non-genetic etiologies, the finding of ataxia may be a single disease or a component of complex diseases. At the same time, the onset time and variety of the disease may differ even among the members of the same family. In this study, the variability of genetic etiology and phenotype in patients with ataxia symptoms is presented. The power of next-generation sequencing analysis, such as WES and CES, in detecting the etiology of patients with ataxia of unknown cause was evaluated. Informed consent forms were obtained from all patients. Data usage approval was taken from Afyonkarahisar Health Sciences University Ethics Board.
Results
Demographical data, family history, and clinical findings of 36 patients included in the study are summarized in Table 1. Based on the table, the onset age of the disease in 15 of the patients is 18 years or younger. In 21 of them, the symptoms started after the age of 18. Parents of 66.6% of the cohort had consanguineous marriage, while 27.7% did not describe consanguinity between their parents, but they were from the same village. According to the physical examination, 77.7% of the patients had dysmetria, 66.6% had dysdiadokinesia, and 61.1% had dysarthria.Table 1. Summary of patients’ demographic, family, and clinical findingsPatient ID#1#2#3#4#5#6#7#8SexFMFMMFFMAge at diagnosis (Y)2818234648482341Age at onset (Y)1820211214402040Consanguineous marriage + + + + + ---1st-degree cousin + + + 2nd-degree cousin + + 3rd-degree cousinFrom same village + + Other affected individuals in familyNoNo1 cousin1 brother (patient #6)1 brother (patient #5)GrandmotherNoNoClinical featuresAtaxia + + + + + + + + Dysmetria + - + + Could not be evaluated due to hearing loss + + + Dysdiadochokinesia-- + + Could not be evaluated due to hearing loss + + + Dysarthria + + - + + - + + OthersHeadache, bilateral horizontal and vertical nystagmus, mild tongue fibrillation, bilateral pes cavusDifficulty swallowing, muscle weakness, microcephaly, neuromotor development delay (in childhood), hyperreflexia, bilateral ptosisSeizure, tremor, hyperreflexia, mild mental retardationSpastic tetraparesis, dysphagia, hyperreflexia, bilateral pes cavus, bilateral cataract, bilateral sensorineural hearing lossSpastic paraparesis, hyperreflexia, bilateral pes cavus, bilateral cataract, bilateral sensorineural hearing lossHead titubation, tremor, hyperreflexia, cercival dystonia, mixt type incontinenceHeadache, bilateral horizontal nystagmus, urinary incontinence, hyperresflexia, inability to walk independently, bilateral pes cavus, secondary amenorrheaPlantar reflex indifference, romberg positive, decreased lower extremity deep sensation, pes cavus, hammertoes, nistagmus, frequent falls#9#10#11#12#13#14#15#16#17MMMMMMFFM195025345764282229Infant period171629In childhood5371922- + + + -- + + + + + + + + + + + + NoGrandson of uncle’s sonNoNoSisterNoBrotherBrother and paternal uncleUncle + + + + (1 day a week) + + + + + + + - + + + + + + + + - + + + - + + - + - + + + - + + Mental motor retardation, divergent strabismus, mild muscle weakness, hyperreflexia, poor eye contact, ticsTremor, myoclonus, epilepsy, mild facial muscle weaknessMild intellectual disability, pes cavus, muscle weakness in the lower and upper extremities, hyperreflexia, spasticity in the lower extremitiesHorizontal and vertical nystagmus, headache, attention deficit, hyperreflexia at lower extremities, tachicardiaHyperreflexia, fibrillation of tongue, intellectual disability, hearing lossNystagmus, mild muscle weakness, spasticity, hyperreflexia, difficulty walking, urinary urgencyMuscle weakness upper and lower extremities, inability to climb stairs, drop foot, decreased deep tendon reflex, loss of feeling in the feet, urinary incontinenceHorizontal nystagmus, decreased deep tendon reflex, tongue fibrillationBilateral hearing loss, epilepsy, mild facial muscle weakness, weakness in hands (bilateral), lordotic posture#18#19#20#21#22#23#24#25MFMMFFFF36515739202034351543494118182033 + + + - + + + + + + + + + + + + NoBrother (patient #22)Sister (patient #21)Brother and motherTwin sister (patient #25)Twin sister (patient #24)NoNo + + + + + + + + - + + + + + + + - + + + + + + + + --- + + - + Inability to walk, pes cavus, hypothenar atrophy, severe muscle weakness in the upper and lower extremities, decreased deep tendon reflex (in 4 extremities)Paraparesis, bilateral pes cavus, spasticity in lower extremities, mild facial muscle weakness, tremor, difficulty walkingParaparesis, hyperreflexia, tremor, difficulty walkingHorizontal nistagmus, muscle weakness, hyperactivity, forgetfulness, behavioral disorder, repetitive speech, seizure, joint pain, urinary incontinenceExercise intolerance, epilepsy, delayed walking and speaking timeEpilepsy, tremor of bilateral hands, delayed walking and speaking timeSevere weakness in ankle dorsiflexion, no global deep tendon reflex, horizontal nystagmus, leg atrophy, bilateral pes cavus, migraine, head titubationMicrocephaly, spastic tetraparesis, hyperreflexia, difficulty walking, difficulty swallowing, amnesia, üriner inkontinans, hepatosplenomegaly, spasticity#26#27#28#39#30#31#32#33#34#35#36MMFMMMMMMMM2339465858491924375842153145475737Childhood period15175130 + + --- + + -- + + + + + + + + + - + + + 2 uncle of sonsGrandson of paternal uncleSisterBrotherBrotherPaternal uncle’s daughterNoNoDaughterDaughter, 4 nephews, father, uncle, paternal grandmotherSister, mother + + + + + + + + + + + + + - + + + + --- + + + -- + + + ----- + - + + + - + + --Inability to walk, decreased deep tendon reflex, difficulty in dorsiflexion in feet, atrophy of legs, arms and facial musclesHyperreflexia, facial asymmetry, limitation of eye movements to look downDementia, severe cognitive impairment, hydrocephaly, hyperreflexiaBilateral glaucoma, horizontal nystagmus, tongue atrophy, decreased global deep tendon reflex, thenar and hypothenar atrophy, proximal muscle weaknessMuscle weakness in the hands and feet, urinary incontinence, difficulty walking, numbness in the shoulder, increased deep tendon reflex in the upper extremitiesTongue fibrillation, muscle weakness, upward gaze limitation, ptosis, difficulty walking, spasticity, hyperreflexiaGrowth retardation, microcephaly, nistagmus, unilateral renal atrophy, delayed puberty, hyperreflexia, conical teethSpasticity, urinary incontinence, difficulty walking, paraplegia, hyperrefleksi, bilateral plantar reflex flexionWeakness in the feetBilateral plantar reflex indifference, global hiperrefleksia, decreased sense of vibration in the lower extremities, spasticity in the lower extremity, pain in your kneesBilateral ptosis, limited outward gaze in both eyes, difficulty in swallowing, nazone speech, bilateral upper extremity proximal muscle atrophy, muscle weakness, dysphagia, hyporeflexia
Brain MRI, electromyography, laboratory, and other test results of the patients are presented in Table 2. Thirty-three of the patients had brain MRI images and 26 of them had abnormal cerebellum image (mostly atrophy). In addition, 20 of them had EMG results and 10 of them had polyneuropathy.Table 2. Summary of patients brain MR images, electromyography, and biochemical test resultsPatient IDBrain MRI findingsElectromyography resultsLabOther#1Mild cerebellar atrophy, diffuse white matter damageN/A--#2NormalNormalNormalEndoscopy: erythematous gastropathy#3Cerebellar atrophyN/ADecreased serum folate (result 2.17 µg/L)-#4N/ASensorineural polyneuropathy--#5N/AN/A-Perforation of the stomach history#6Cerebral hemisphere atrophy, cystic encephalomalacia in both cerebellar hemispheres, T2 hypointense signal change due to degenerative ion accumulation in bilateral globus pallidusN/A--#7Cerebral and cerebellar hemisphere atrophy, vanishing white matter (more prominent at the level of the corona radiata and centrum semiovale)NormalFSH and LH increased, estradiol decreasedAbdominal ultrasonography: uterine size is small. Bilateral ovaries were not seen clearly#8Ischemic focus in bilateral cerebral parenchyma, mild cerebellar atrophyDemyelinating type polyneuropathyACTH decrease, cortisol decreaseIsolated adrenocorticotropic hormone deficiency#9Cerebellar hypoplasia, hypoplasia of the cerebellar vermis, superior cerebellar peduncle elonged and thinned (molar tooth sign)N/A--#10Cerebellar atrophyN/A-EEG: normal#11Hyperintensity in periventricular and supraventricular white matterChronic motor axonal polyneuropathy--#12Superior cerebellar atrophyBilateral mild carpal tunnel syndrome-ECG: sinusal tachhycardia, long PR interval ECG Holter:bigemine bets. Supraventricular tachicardic attacks present echo: ef 60% tachicardic#13Minimal cerebral cortical atrophy, severe cerebellar atrophyPolyneuropathy--#14Cerebral cortical atrophy, diffuse cerebellar atrophy, prominent cerebellar foils, degenerative changes in the ponsDemyelinating sensorimotor polyneuropathy--#15Mild cerebellar atrophyMotor axonal polyneuropathyMild elevated CK, proteinuria, hematuriaObesity, gestational diabetes#16Cerebellar cyst and atrophy of the cerebellum anteriorN/A--#17Mild cerebral and cerebellar atrophyN/A-Hypertension, diabetes, bilateral glaucoma#18NormalMotor neuropathyCK: 243-#19Cerebral and cerebellar atrophyN/A--#20Cerebral and cerebellar atrophyN/A--#21Dilatation of both lateral ventricles (hydrocephalus), gliosis of periventricular and subcortical deep white matterN/A--#22Mild cerebellar atrophyN/A-EEG: focal epileptic anomaly#23Mild cerebellar atrophyN/A-EEG: focal epileptic anomaly#24Atrophy of both cerebellar hemispheres, lateral and third ventricles and fourth ventricle and folios prominently enlargedDemyelinating sensorimotor polyneuropathy--#25Cerebral cortical atrophy, hyperintensity in bilateral periventricular white matter, corona radiata level hyperintensity in right frontal deep white matterNormal-Abdominal USG: hepatosplenomegaly#26Cerebellar atrophy, prominence of cerebellar foils, mega cisterna magnaCompatible with myopathyCK: 650-#27Volume loss in both cerebellar hemispheres and vermis, cerebellar foils prominent, hyperintensity in periventricular white matter, mesencephalon atrophicN/A--#28Diffuse cerebral and mild cerebellar atrophy, hyperintensity in supratentorial deep white matterN/A-Mini Mental test result 6/30#29Cerebellar atrophy, prominence of cerebellar foils, cerebral atrophyDemyelinating sensorimotor polyneuropathyIncreased lactate dehydrogenase (474–4685)-#30Mild cerebral cortical atrophy, mild cerebellar atrophyN/A-Hypertension#31Cerebellar atrophy, prominence of cerebellar foils, cerebral atrophyNormal-EEG: normal#32Basilar invagination, hyperintensity in the periventricular white matter, diffuse thinning of the corpus callosum, narrowing of the foramen magnum, pencil-like ending at the lower end of the cerebellumNormalDecreased testosteroneSomatosensory evoked potential: bilateral tibial SEP cortical response could not be obtained. Cervical MR: at C1 vertebra level, centrally located. Scrotal USG: left testis was not observed. Right testis atrophicfocal signal increase in the spinal cord draws attention. It was evaluated in favor of myelomalastic changes#33Thin corpus callosumNormalCK: 209Muscle biopsy: loss of myelin (dysferlin)#34Hyperintensity in both centrum semiovale posterior and both cerebellar hemispheres with patchy and occasionally nodular configurationPolyneuropathy involving motor fibers--#35Moderate cerebral atrophyBilateral moderate carpal tunnel syndrome with diabetic polyneuropathyHypertension, diabetes mellitus#36N/ANormalIncreased CK (2119)Abdomen USG: grade 2 steatosis
WES analysis was performed on all patients. In total, 36 patients had 38 different variants. Two different variants were found in 6 of 37 patients (patient numbers 4, 12, 13, 15, 18, and 20). In addition, the same variants were present in 3 pairs of siblings in the cohort (patient numbers 5 and 6, 19 and 20, and 22 and 23). The variants compatible with the phenotypes of the patients with autosomal dominant inheritance are presented in Table 3, and those with autosomal recessive inheritance are presented in Table 4. Patient #17 is not included in both tables. In the WES analysis of this patient, no point mutation compatible with the phenotype was found. The Franklin genetic analysis program provides CNV (copy number variations) analysis in the WES. When CNV analysis was performed for this patient, approximately 3.5 kilobases of homozygous deletion was detected at coordinate chr11:118.895.610–118.899.076. Other genetic diagnostic methods subsequently confirmed this deletion for this patient. Homozygous mutations of the TRAPPC4 gene located between these regions cause autosomal recessive inheritance disease “Neurodevelopmental disorder with epilepsy, spasticity, and brain atrophy—NEDESBA.” The phenotype of the patient is compatible with this disease.Table 3. Genetic results of patients with autosomal dominant inheritance diseasePatient IDGenesZygosityVariantMutation typeID (NM!!)gnomAD FREQUENCY (Aggregated—Aggregation of gnomAD exome + genome-)AGMG classification/pathogenicity criteria (ACMG)Genetic diagnosisOMIM#10KCNC3Heterozygousc.124C > TNonsenseNM_004977.2N/ALikely pathogenic/PVS1, PM2, PM6Spinocerebellar ataxia 13#605259#12CACNA1AHeterozygousc.5248C > TMissenseNM_001127222.1N/ALikely pathogenic/PM2, PP3, PP2, PP5Episodic ataxia, type 2#108500#12SCN2AHeterozygousc.5011A > GMissenseNM_001040142.2N/ALikely pathogenic/PM1, PP2, PM2, PP3Episodic ataxia, type 9#618924#18PRKCGHeterozygousc.1381G > AMissenseNM_002739.5 < 0.01Likely pathogenic (ClinVar Likely Pathogenic)/PM2, PP2, PP5, PM6Spinocerebellar ataxia 14#605361#28NF1Heterozygousc.4246C > TMissenseNM_001042492.2N/ALikely pathogenic/PM1, PP2, PM2, PP3Neurofibromatosis, type 1#162200#30SH3TC2Heterozygousc.2491_2492delAGFrameshiftNM_024577.3 < 0.01Likely pathogenic/PVS1, PM2Mononeuropathy of the median nerve, mild#613353#34CACNA1AHeterozygousc.4471G > AMissenseENST00000360228.5 < 0.01Likely pathogenic/PM1, PP2, PM2, PP3Episodic ataxia, type 2#108500#35SPASTHeterozygousc.1567_1567delTFrameshiftNM_014946.3N/ALikely pathogenic/PVS1, PM2Spastic paraplegia 4, autosomal dominant#182601#36POLGHeterozygousc.3293A > CMissenseNM_001126131.2N/ALikely pathogenic (ClinVar VUS)/PM1, PP2, PM2, PP3, PM5Progressive external ophthalmoplegia, autosomal dominant 1#157640Table 4Genetic results of patients with autosomal recessive inheritancePatient IDGenesZygosityVariantsMutation typeID (NM!!)gnomAD FREQUENCY (Aggregated—Aggregation of gnomAD exome + genome-)Franklin AGMG classification/pathogenicity criteria (ACMG)Genetic diagnosisOMIM#1AGTPBP1Homozygousc.436 + 4A > GSplicing variantNM_001330701.2 < 0.01%VUS/PM2, PP3Neurodegeneration, childhood-onset, with cerebellar atrophy#618276#2SLC52A2Homozygousc.889G > AMissenseNM_001363118.2 < 0.01%VUS/PM2, PM1Brown-Vialetto-Van Laere syndrome 2#614707#3FOLR1Homozygousc.640_676delTTCGACCCAGCCCAGGGCAACCCCA ATGAGGAGGTGGFrameshiftNM_016729.3N/ALikely pathogenic/PVS1, PM2Neurodegeneration due to cerebral folate transport deficiency#613068#4ABHD12Homozygousc.1124_1129delTTTACAIn frame deletionNM_001042472.3N/ALikely pathogenic/PP1, PM2, PM4, PP5Polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataract#612674#5ABHD12Homozygousc.1124_1129delTTTACAIn frame deletionNM_001042472.3N/ALikely pathogenic/PP1, PM2, PM4, PP5Polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataract#612674#6AGTPBP1Homozygousc.2969A > TMissenseNM_001330701.2N/AVUS (ClinVar Pathogenic)/PM2, PP5Neurodegeneration, childhood-onset, with cerebellar atrophy#613068#7EIF2B3Homozygousc.179_187delAAAAGGCTCIn frame deletionNM_001330701.1N/AVUS/PP1, PM2, PM4,Leukoencephalopathy with vanishing white matter#603896#8SACSHomozygousc.13346G > TMissenseNM_014363.6N/AVUS/PM2, PP3Spastic ataxia, Charlevoix-Saguenay type#270550#9ATCAYHomozygousc.552C > GNonsenseNM_033064.5N/ALikely pathogenic/PVS1, PM2Ataxia, cerebellar, Cayman type#601238#11ZFYVE26Homozygousc.2263C > TNonsenseNM_015346.4N/APathogenic/PVS1, PM2, PP5Spastic paraplegia 15, autosomal recessive#270700#13GDAP2Homozygousc.757C > TNonsenseNM_017686.4 < 0.01%Pathogenic/PVS1, PM2, PP5Spinocerebellar ataxia, autosomal recessive 27#618369#13PLA2G6Homozygousc.1063C > TMissenseNM_003560.3 < 0.01%VUS (ClinVar Uncertain significance)/PM2, PP3, PP2Neurodegeneration with brain iron accumulation 2B#610217#14SPG7Homozygousc.233T > ANonsenseNM_003119.40.04%Pathogenic/PVS1, PM2, PP5Spastic paraplegia 7, autosomal recessive#607259#15LPIN1Homozygousc.151C > TMissenseNM_001349206.1 < 0.01%Likely pathogenic/PP3, PM2Myoglobinuria, acute recurrent, autosomal recessive#268200#15MCM3APHomozygousc.1226T > CMissenseNM_003906.50.03%VUS (ClinVar Uncertain significance)/PM2Peripheral neuropathy, autosomal recessive, with or without impaired intellectual development#618124#16SETXHomozygousc.5549-2A > GSplicing variantNM_ 015046.7N/ALikely pathogenic/PVS1, PM2Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 2#606002#18SYNE1Homozygousc.18658C > AMissenseNM_182961.4 < 0.01%VUS (ClinVar Uncertain significance)/PM2Spinocerebellar ataxia, autosomal recessive 8#610743#19KIF1CHomozygousc.328C > TNonsenseNM_006612.6N/APathogenic (ClinVar Pathogenic)/PVS1, PM2, PP5Spastic ataxia 2, autosomal recessive#611302#20KIF1CHomozygousc.328C > TNonsenseNM_006612.6N/APathogenic (ClinVar Pathogenic)/PVS1, PM2, PP5Spastic ataxia 2, autosomal recessive#611303#21TREM2Homozygousc.197C > TMissenseNM_018965.3 < 0.01%Likely pathogenic (ClinVar Pathogenic)/PM2, PP5Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy 2#618193#22COQ8AHomozygousc.815G > CMissenseNM_020247.5N/ALikely pathogenic/PP1 (MANUEL), PM2, PM5, PM1, PP3Coenzyme Q10 deficiency, primary, 4#612016#23COQ8AHomozygousc.815G > CMissenseNM_020247.5N/ALikely pathogenic/PP1 (MANUEL), PM2, PM5, PM1, PP3Coenzyme Q10 deficiency, primary, 4#612016#24SETXHomozygousc.5549-2A > GSplicing variantNM_015046.7N/ALikely pathogenic/PVS1, PM2Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 2#606002#25NPC1Homozygousc.3019C > GMissenseNM_000271.50.02%Pathogenic (ClinVar Pathogenic)/PM1, PP2, PM2, PM5, PP3, PP5Niemann-Pick disease, type C1/D#257220#26TTPAHomozygousc.205-1delGSplicing variantNM_000370.3N/ALikely pathogenic/PVS1, PM2Ataxia with isolated vitamin E deficiency#277460#27NPC1Homozygousc.1421C > TMissenseNM_000271.5 < 0.01%Pathogenic (ClinVar Pathogenic)/PM2, PM5, PM1, PP2, PP3, PP5Niemann-Pick disease, type C1/D#257220#29APTXHomozygousc.884C > TMissenseNM_001195248.2N/AVUS (ClinVar Uncertain significance)/PM2, PP3Ataxia, early-onset, with oculomotor apraxia and hypoalbuminemia#208920#31AFG3L2Homozygousc.2044G > TMissenseNM_006796.2N/AVUS/PM2, PM1, PP2Spastic ataxia 5, autosomal recessive#614487#32PEX10Homozygousc.194-6T > CSplicing variantNM_153818.1N/AVUS/PM2, BP4Peroxisome biogenesis disorder 6B#614871#33SPG11Heterozygousc.1235C > GNonsenseNM_025137.4 < 0.01%Pathogenic (ClinVar Pathogenic)/PVS1, PM2, PP5Spastic paraplegia 11, autosomal recessive#604360Heterozygousc.1699C > TNonsenseNM_025137.4N/ALikely pathogenic/PVS1, PM2
Discussion
This study presented the genetic results of patients who were referred to medical genetics and had ataxia symptoms. Genetic testing should be considered in patients in whom an organic cause for ataxia cannot be found through current diagnostic methods. In addition, finding of the genetic cause for complex diseases with ataxia component provides a holistic approach in follow-up.
In the literature review, the prevalence of hereditary was found to be higher than autosomal recessive inheritance ataxias in autosomal dominant inheritance [8, 9]. In our study, autosomal recessive inheritance was found in 28 of 36 patients, and autosomal dominant inheritance was found in 9 patients. High rate of consanguineous marriages in our region (66.6% consanguineous marriage and 27.7% parents from the same village) was interpreted as the reason behind the higher detection of patients with autosomal recessive inheritance. In a similar study conducted by Galatolo et al., similar to our results, recessive genetic diseases were detected more frequently than dominant diseases in ataxia patients [10]. The age of onset of the disease in 41.6% of patients was before the age of 18. In a similar study conducted by Coutelier et al., the age of onset of the disease was younger than 25 years in 44% of the patients. This situation was found to be similar to our study [11]. Thirty-eight different variants were detected. Among them, 12 were VUS (1 autosomal dominant and 11 autosomal recessive), 19 were likely pathogenic (9 autosomal dominant and 10 autosomal recessive), and 8 were pathogenic (7 autosomal recessive). None of VUS variants were in the autosomal dominant group, while 11 of them were in the autosomal recessive group. We think that the possible reason for this is that autosomal recessive diseases are seen less frequently in the population and therefore fewer cases are reported.
The most common brain MRI result, reported in patients with ataxia, is cerebellar atrophy. Other cerebellar changes and atrophy of brain cortex, brain stem, striatum, and spinal cord have also been reported [12, 13]. In our study, 2 of the 36 patients with brain MRI results were normal. Patient #25 and patient #27 are patients diagnosed with “Niemann-Pick disease, type C1 / D,” carrying homozygous variants in the NPC1 gene. Although behavioral changes are usually reported as the first symptom in these patients, the first findings in our patients were cerebellar findings due to cerebellar atrophy [14] (Fig. 1). Additional tests for intracellular cholesterol homeostasis in cultured fibroblasts, “Sea-blue” histiocytes appearance, and biochemical tests are planned for the patients. It should be taken into consideration that metabolic diseases can also be seen in adult patients, and clinical findings may be different compared to pediatric patients. All radiological and biochemical tests performed on patients are important clues in the approach from phenotype to genotype. Patient #28 was referred to us with findings of ataxia, dementia, and severe cognitive impairment. Additionally, the patient had ataxia for 1 year as widespread cerebral and cerebellar atrophy had developed. As a result of the WES analysis, a likely pathogenic heterozygous variant was detected in the NF1 gene. Thereupon, the patient was re-evaluated following the genotype-to-phenotype approach. The patient did not have the necessary findings for “Neurofibromatosis, type 1” diagnosis. In 2021, Kallionpää et al. reported an increased risk of dementia in NF1 patients [15]; thus, dementia and severe cognitive impairment findings of the patient were considered as possible results of this mutation. As a result of WES analysis of patient #30, a heterozygous likely pathogenic variant was detected in the SH3TC2 gene. The resultant clinical findings are not fully responsible for “Mononeuropathy of the median nerve, mild.” Cerebellar findings are prominent in the patient’s clinic. Homozygous variants in the SH3TC2 gene cause Charcot-Marie-Tooth disease, and cerebellar atrophy has been reported in 20% of patients [16]. A definitive diagnosis could not be made for this patient, and two possible options were considered. (1) There is a different variant on the other allele in the patient’s somatic tissue (especially the central nervous system), and, therefore, the CMT clinic remains mild, but cerebellar findings are apparent. (2) The detected variant is actually responsible for the clinic, but there are no reported cases yet.Fig. 1A, B Brain MRI image of patient #25 (sagittal and coronal). C, D Brain MRI image of patient #27 (sagittal and coronal). Both patients have a homozygous variant in the NPC1 gene and were diagnosed with “Niemann-Pick disease, type C1/D.”
Multiple involvement is expected in long-term diseases such as neurological diseases. In this case, all examinations such as EMG, as well as the patient’s MRI image, become important. In addition, some diseases can manifest themselves in different clinical components like ataxia and polyneuropathy. In our study, EMG was performed on 20 of 36 patients with ataxia, and polyneuropathy was detected in 10 of them. Many diseases accompanied by ataxia and polyneuropathy have been reported, and EMG is recommended in ataxias of unknown etiology [17–19].
Two variants one patient
WES and CES analyze thousands of genes that cause thousands of diseases. According to the results, a patient may sometimes have variants that can cause more than one disease [20]. The outcome report in this study presented two different variants that may cause disease in six patients (patient numbers 4, 12, 13, 15, 18, and 20).
WES analysis of patient #13 detected homozygous variants in GDAP2 and PLA2G6 genes. The variant detected in the GDAP2 gene is pathogenic according to ACMG classification and causes “Spinocerebellar ataxia, autosomal recessive 27” disease. The clinical findings of the patient were thought to be compatible with this disease. Clinical signs of the patient started in childhood. However, the symptoms started over the age of 30 in the reported cases [21]. In addition, while dementia was present in these reported cases, our case had an intellectual disability present since childhood. The variant in the PLA2G6 gene detected as a result of WES was classified as VUS according to ACMG. This variant was previously reported in the ClinVar database (SCV001992003). Three different diseases caused by the variant in this gene have been reported in the OMIM database. Among these three diseases, it was considered that “Neurodegeneration with brain iron accumulation 2B—Atypical neuroaxonal dystrophy” could be compatible with the patient’s clinic. The intellectual disability and the onset of speech and gait problems in childhood are consistent with this disease [22]. However, the MR images of the patient are not compatible with the MR images reported in the disease [23]. Considering the onset age of the patient’s symptoms and the signs of intellectual disability, the condition could be compatible with “Atypical neuroaxonal dystrophy” [24]. In addition, the variant detected in the GDAP2 gene was considered to be responsible for the patient’s phenotype like cerebellar atrophy in brain MRI. However, it differed from the cases reported in the literature in that he had much earlier onset. It was thought that both variants of this patient’s phenotype could be responsible. The overlap of some clinical findings of the patient with both diseases caused them to be considered in the diagnosis.
Another patient with two different variants is patient #15. Homozygous variants in the LPIN1 gene and MCM3AP gene were detected in this patient. Homozygous variants in the LPIN1 gene cause “Myoglobinuria, acute recurrent, autosomal recessive” disease. When the patient was questioned again after obtaining the WES result, it was learned that she had recurring mild elevated CK, proteinuria, and hematuria since childhood and is being followed up by the nephrology department. The present nephrological findings of the patient were considered because of this variant [25]. Variants in the MCM3AP gene cause “Peripheral neuropathy, autosomal recessive, with or without impaired intellectual development.” Ataxia, dysarthria, muscle weakness, hyporeflexia, drop foot, loss of ambulation, loss of sensation in the feet, obesity, and motor axonal polyneuropathy on EMG were detected in the patient. The variant in the MCM3AP gene is thought to be responsible for these clinical findings of the patient. The onset of clinical findings at the age of seven was also considered important [26, 27]. In addition, intellectual disability was reported in the patients, which was not present in our case [27]. In our literature review, increased signal intensity in temporal lobes, mild ventriculomegaly, and white matter cysts were reported in the MRI image, but cerebellar atrophy was not reported in this patient group. Observance of mild cerebellar atrophy in the MRI image of our patient [27] suggested that this variant could be responsible for cerebellar atrophy.
In this study, two different variants compatible with the phenotype, both autosomal dominant inheritance (PRKCG c.1381G > A) and autosomal recessive inheritance (SYNE1 c.18658C > A), were detected in one patient (#18). The patient who has ataxia, pes cavus, dysarthria, impaired cognition, onset at ages 6–40, and motor neuropathy on EMG suggests that it is compatible with “Spinocerebellar ataxia, autosomal recessive 8” caused by homozygous mutation in the SYNE1 gene [28]. Also, likely pathogenic variant in the PRKCG gene detected in this patient was previously reported in ClinVar. Heterozygous variants in this gene cause “Spinocerebellar ataxia 14” disease. The patient’s ataxia, dysarthria, and cognitive impairment symptoms are compatible with the disease [29]. However, the finding of motor neuropathy on EMG and the onset of the disease at the age of 20 are incompatible with this disease. In addition, the patient’s brain MR image was reported as normal. In this case, while the neuropathy findings of the patient were compatible with the variant in the SYNE1 gene, the variant in the PRKCG gene could not be ruled out and it is thought to be effective on the phenotype.
Patient #19 and patient #20 are siblings with similar clinical findings. A pathogenic homozygous change in the KIF1C gene was detected in both patients. The clinical findings are compatible with “Spastic ataxia 2, autosomal recessive” caused by variants in the KIF1C gene. Literature reported that the symptoms in this group of patients start in their teens [30, 31]. However, the onset age of the symptoms of our patients was in their forties. Cerebellar atrophy is not a very common finding in these patients, and cerebellar atrophy was present on brain MRI images in both of our patients [30] (Fig. 2). Additionally, a heterozygous pathogenic variant in the CCDC88C gene was detected in patient #20, unlike his sister. The disease “Spinocerebellar ataxia 40” caused by this variant matches the clinical findings of patient #20. At this point, it is very difficult to pinpoint the variant responsible for his phenotype. A good anamnesis explaining the natural history of the disease in detail is important at this point. These two siblings have exactly the same anamnesis and similar clinical findings. In this case, their sharing of the same mutation like KIF1C gene variant is considered to be more likely.Fig. 2A, B Brain MRI image of patient #19 (sagittal and coronal). C, D Brain MRI image of patient #20 (sagittal and coronal). Diffuse cerebral and cerebellar atrophy were present in both patients
Treatment option
Patient #12 was referred to our department with signs of nystagmus, ataxia (episodic—one day a week), headache, attention deficit, and increased deep tendon reflex in the lower extremities. Also, the 8-year-old son of the proband has speech problem (like dysarthria) and balance disorder. Two different variants were found in the WES analysis of the patient (“CACNA1A gene c.5248C > T as heterozygous” and “SCN2A gene c.5011A > G as heterozygous”). Since these two variants were classified as likely pathogenic, they were considered to be responsible for the patient’s phenotype. According to the OMIM database, variants in the CACNA1A gene cause “Developmental and epileptic encephalopathy 42,” “Episodic ataxia, type 2,” “Migraine, familial hemiplegic, 1,” “Migraine, familial hemiplegic, 1, with progressive cerebellar ataxia,” and “Spinocerebellar ataxia 6” diseases. The clinical findings of the patient were considered to be compatible with “Episodic ataxia, type 2.” In the family history information obtained from the patient, no individual with similar clinical findings was found. There may be two reasons for this situation; first, it may be a de novo variant; second, incomplete penetrance may be the case for this family [32]. Variants in the SCN2A gene cause three diseases according to the OMIM database: “Developmental and epileptic encephalopathy 11,” “Episodic ataxia, type 9,” and “Seizures, benign familial infantile, 3.” Since the 34-year-old patient had no history of seizures, the condition was thought to be compatible with “Episodic ataxia, type 9.” In this disease, most patients have a history of seizures [33, 34]. In the study by Schwarz et al., seizures were not reported in 3 of 21 patients with SCN2A-associated episodic ataxia. The age of onset of ataxia in these 3 patients reported variances between 1 and 3 years [35]. In our case, the onset age of ataxia was 29. Since the variant detected in the patient was likely pathogenic, it was considered that the variant could be responsible for the phenotype. However, the absence of epilepsy and the late onset of ataxia differed our case from others reported in the literature. In addition, acetazolamide is used in the treatment of both diseases [36]. In this patient, acetazolamide treatment was decided based on the WES result. It is an exemplary case showing the significance of WES test in medical treatment.
CNV results
Patient #17 is a good example of the tests being developed considerably for genetic diagnosis in recent years. The WES analysis performed on the patient detected approximately 3.5 kilobases of homozygous deletion. This deletion involving the TRAPPC4 gene is consistent with the patient’s clinical findings. This patient is a good example for large CNV analysis with WES [37, 38]. Over time, it will become possible to detect all kinds of mutations with a single molecular genetic analysis such as WES or similar.
Mild phenotype
The homozygous variants in the AGTPBP1 gene detected in patient #1 and patient #6 constitute another result that is different from the literature. Homozygous variants in this gene cause “Neurodegeneration, childhood-onset, with cerebellar atrophy.” Although the known symptoms of the disease are severe, they appear in infancy and death occurs in childhood [39]. Our patients have ataxia accompanied by different neurological findings. Definitive genetic diagnosis was not possible for these two patients. In the future, the literature may report that homozygous variants in the AGTPBP1 gene cause a disease with a milder clinical condition.
Limitations
The major limitations of the study are that we cannot detect some variants like trinucleotide repeat expansions that cannot be detected by WES nor CES. This can be avoided by choosing another sequencing approach.
Conclusions
For neurological diseases with unknown etiology and treatment today, medical genetic aspect is like a light at the end of a dark tunnel. A variety of technologies and tests have been developed for diagnosis in medical genetics. New genetic tests have recently become more important in the ataxia patient group with an undeterminable etiology. With the development of technology, the increasing power of genetic diagnosis has enabled the diagnosis of patients with clinical findings not yet fully established. If there are individuals in the family with the same clinical findings or if parents had a consanguineous marriage constituting a clue for recessive diseases, a genetic cause should be considered. The diagnosis of ataxia patients with unknown etiology, which can be described, is made possible thanks to these clues.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Millan F, Mc Knight D, Scuffins J, Juusola J, Richard G, Lindy A (2017) Exome sequencing offers a comprehensive genetic evaluation and high diagnostic rate for ataxia-related disorders (S 17.004). Neurology 88:S 17.004. http://n.neurology.org/content/88/16_Supplement/S 17.004.abstract
- 2Coutelier M, Hammer MB, Stevanin G, Monin M-L, Davoine C-S, Mochel F, Labauge P, Ewenczyk C, Ding J, Gibbs JR, Hannequin D, Melki J, Toutain A, Laugel V, Forlani S, Charles P, Broussolle E, Thobois S, Afenjar A, Anheim M, Calvas P, Castelnovo G, de Broucker T, Vidailhet M, Moulignier A, Ghnassia RT, Tallaksen C, Mignot C, Goizet C, Le Ber I, Ollagnon-Roman E, Pouget J, Brice A, Singleton A, Durr A, for the S.P. and A. Network (2018) Efficacy of exome-targeted capture sequencing to detect mutatio · doi ↗ · pubmed ↗
- 3Gwathmey KG, Pearson KT (2019) Diagnosis and management of sensory polyneuropathy. BMJ l 1108. 10.1136/bmj.l 110810.1136/bmj.l 110831068323 · doi ↗ · pubmed ↗
- 4Hogarth P (2015) Neurodegeneration with brain iron accumulation: diagnosis and management. J Mov Disord 8. 10.14802/jmd.14034/J 10.14802/jmd.14034 PMC 429871325614780 · doi ↗ · pubmed ↗
- 5Gregory A, Kurian MA, Maher ER, Hogarth P, Hayflick SJ (1993) PLA 2G 6-associated neurodegeneration. University of Washington, Seattle, Seattle (WA). http://europepmc.org/abstract/MED/2030171820301718 · pubmed ↗