Integrative Transcriptomic and Single-Cell Protein Characterization of Colorectal Carcinoma Delineates Distinct Tumor Immune Microenvironments Associated with Overall Survival
Erika Hissong, Bhavneet Bhinder, Junbum Kim, Kentaro Ohara, Hiranmayi Ravichandran, Majd Al Assaad, Sarah Elsoukkary, Michael Shusterman, Uqba Khan, Kenneth Wha Eng, Rohan Bareja, Jyothi Manohar, Michael Sigouros, Andre F. Rendeiro, Jose Jessurun, Allyson J. Ocean, Andrea Sboner

TL;DR
This study uses advanced techniques to analyze the immune environment in colorectal cancer tumors and finds that immune activity is linked to patient survival.
Contribution
The study identifies two distinct immune microenvironment states in colorectal cancer associated with survival outcomes using integrative transcriptomic and single-cell analysis.
Findings
Two CRC groups were identified: one with an immune-activated phenotype and another with an immunosuppressed microenvironment.
The immunosuppressed group had lower overall survival probability (HR 1.66, p=0.007).
Immune-activated tumors showed higher CD8+ T cells and tertiary lymphoid structures.
Abstract
Colorectal carcinoma (CRC) is a heterogeneous group of tumors with varying therapeutic response and prognosis, and evidence suggests the tumor immune microenvironment (TIME) plays a pivotal role. Using advanced molecular and spatial biology technologies, we aimed to evaluate the TIME in patients with CRC to determine whether specific alterations in the immune composition correlated with prognosis. We identified primary and metastatic tumor samples from 31 consented patients, which were profiled with whole-exome sequencing and bulk RNA-seq. Immune cell deconvolution followed by gene set enrichment analysis and unsupervised clustering was performed. A subset of tumors underwent in situ analysis of the TIME spatial composition at single-cell resolution through Imaging Mass Mass Cytometry. Gene set enrichment analysis revealed two distinct groups of advanced CRC, one with an immune…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
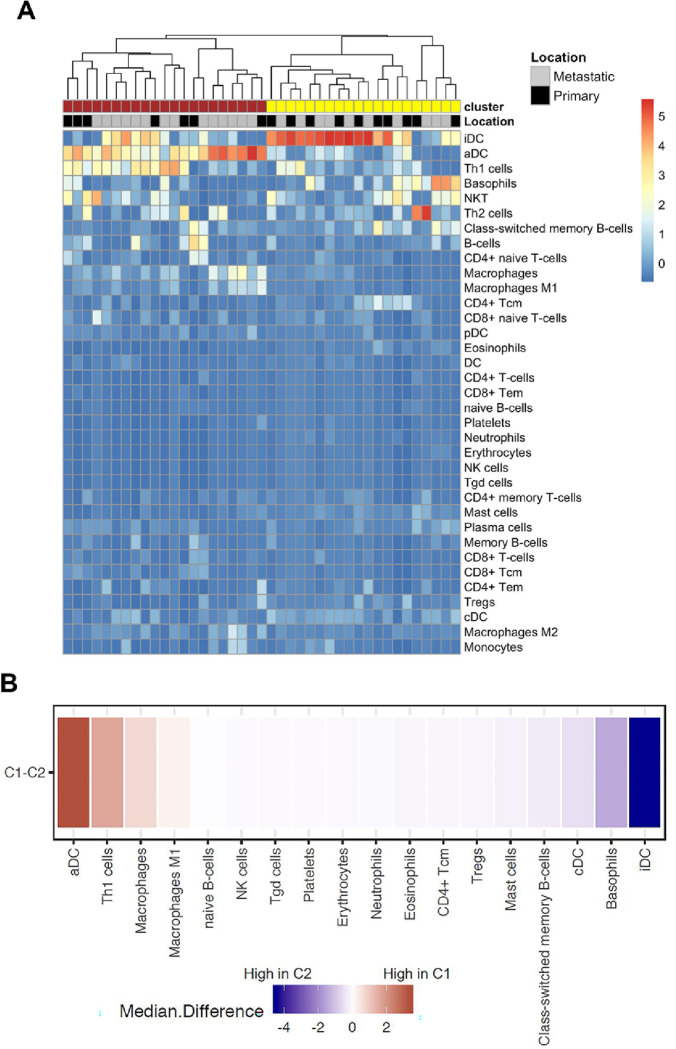 Figure 1
Figure 1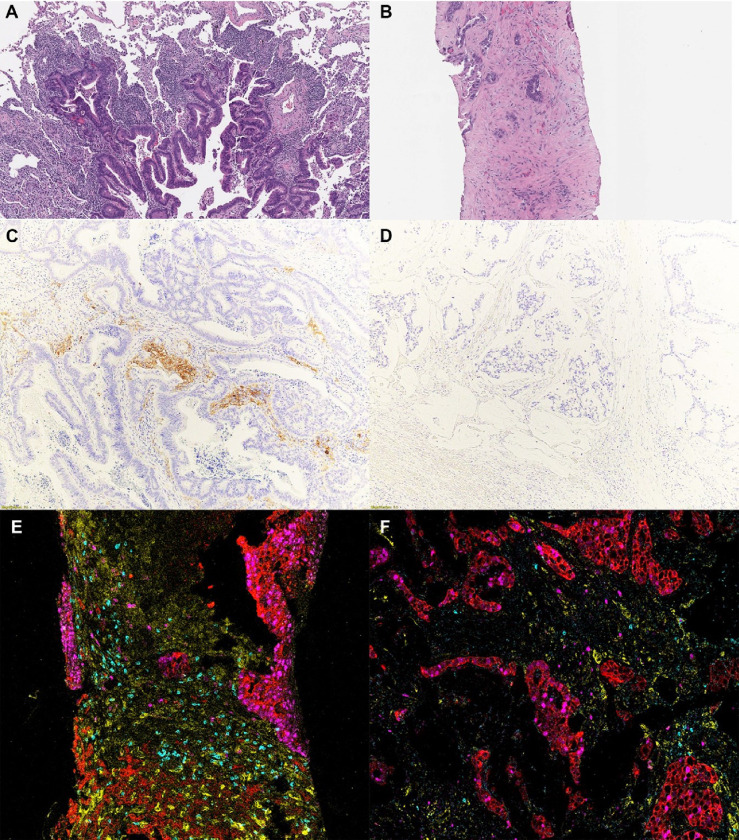 Figure 2
Figure 2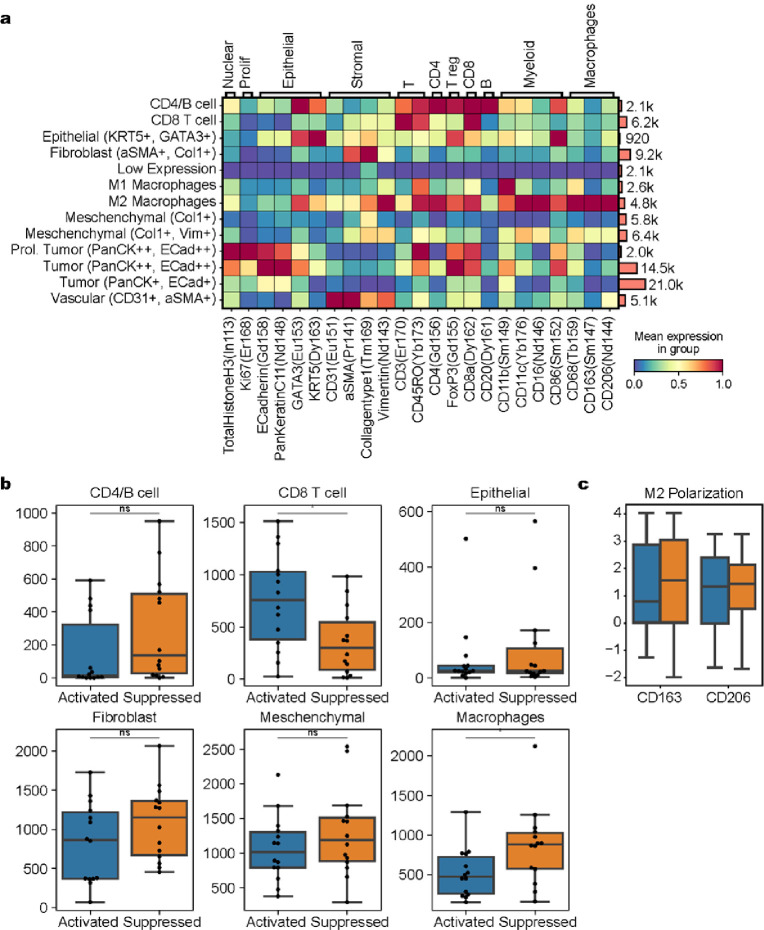 Figure 3
Figure 3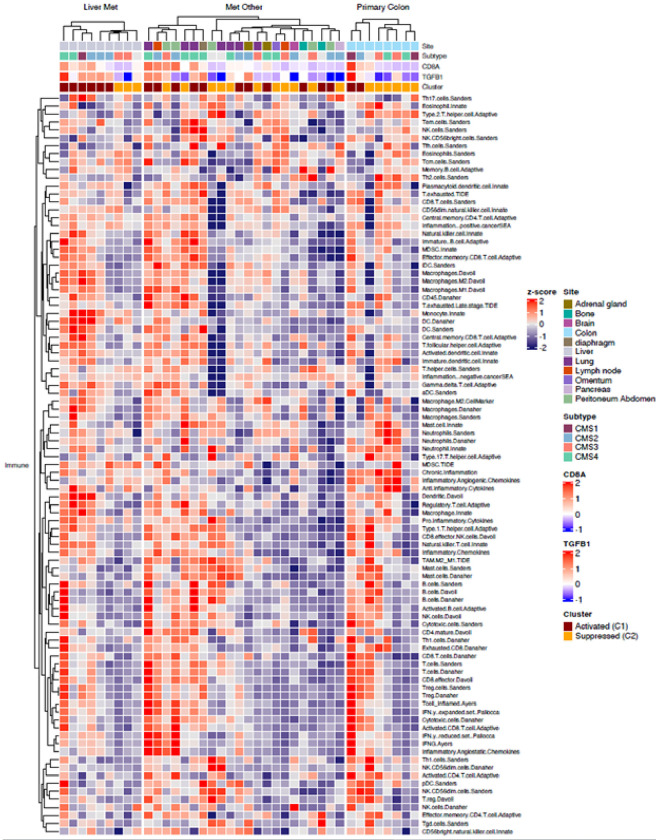 Figure 4
Figure 4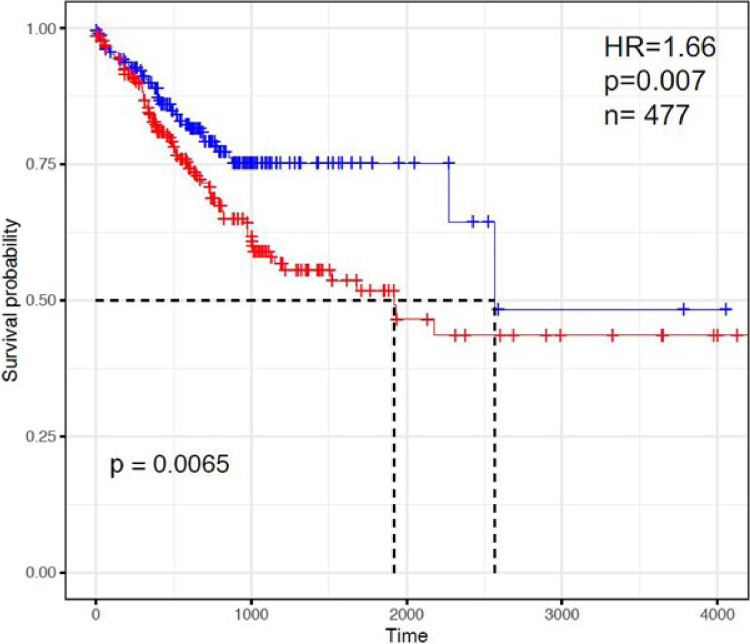 Figure 5
Figure 5- —Englander Institute for Precision Medicine, 2020 Cornell Center for Immunology Core Facilities Seed Grant, NCI
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer Immunotherapy and Biomarkers · Ferroptosis and cancer prognosis · Colorectal Cancer Treatments and Studies
Introduction
Despite advances in screening and early therapeutic interventions, colorectal carcinoma remains a significant cause of morbidity and is one of the leading causes of cancer-related deaths in both men and in women [1]. Up to one-fourth of patients present with metastatic disease at the time of diagnosis and nearly half of patients who have had curable intent resection will still develop recurrence [2–4]. Importantly, colorectal cancer (CRC) is a heterogeneous disease with a diverse molecular background that has significant implications for therapeutic approaches and overall survival. Indeed, unique tumor subsets have specific therapeutic vulnerabilities, such as RAS mutant,mismatch repair deficiency,, or HER2 overexpression.. Advances in the field of molecular pathology have greatly facilitated our understanding of the genomic and epigenomic landscape of CRC, with extensive work done by consortia such as The Cancer Genome Atlas (TCGA) and the international consensus molecular subtype classification consortium, which has enabled the classification of various subtypes of CRC according to their distinct molecular profiles and clinicopathologic features [5,6]. This genomic heterogeneity may partially explain the observation that the aggressiveness of metastatic CRC varies widely among patients, with some harboring localized, resectable metastases that can be managed with curative intent whereas others progress rapidly.
Beyond tumor molecular profiling alone, substantial evidence has highlighted the importance of the tumor immune microenvironment (TIME) in CRC, with some authors suggesting that immune features might serve as a better prognostic indicator than traditional histopathologic methods [7,8]. An example of this, the immunoscore, developed by Galon et al. and based on the quantification of CD3 and CD8-positive T cells using digital imaging analysis, was found to outperform the Tumor-Node-Metastases (TNM) classification for disease-free survival and overall survival and could help predict response to therapy [9–11]. Other studies have found that mature T cells, dendritic cells, and memory T cells were associated with improved prognosis whereas regulatory T cells and M2 macrophages were associated with inferior prognosis [12,13]. Since these initial pivotal studies, our ability to assess the TIME has significantly improved, which has led to a better understanding of its complexity. Recent advances in whole slide scanning, multiplexed single-cell in situ protein profiling and machine learning algorithms have enabled more objective quantification. Additionally, single-cell technologies including RNA sequencing have led to further insights into the TIME [14,15].
The purpose of this study was to evaluate the TIME in patients with CRC to determine whether specific alterations in the immune composition correlated with histopathologic features, molecular alterations, and overall prognosis. We found that CRC TIME could be characterized as either immune activated (higher proportion of activated dendritic cells, tertiary lymphoid structures, higher CD8 + T cells) or inactivated (inactivated dendritic cells, lower CD8 + T cells, increased M2 macrophages), and that the activated immune microenvironment is associated with improved survival.
Materials and Methods
Case selection and histopathology review
We included all patients with colorectal carcinoma who had been consented for molecular testing through the Englander Institute for Precision Medicine at Weill Cornell Medicine [16]. Each sample, including primary and metastatic site, was reviewed by a certified pathologist to confirm the histologic diagnosis. Only fresh frozen samples with sufficient RNA content and quality were included in the study cohort. Clinicopathologic data as well as therapeutic interventions and follow-up information was obtained through review of available surgical pathology material and the electronic medical record. This study was approved by the institutional review board (IRB) at Weill Cornell Medicine (IRB protocol #1305013903).
Whole-exome and bulk RNA sequencing
As described in detail in prior publications [16,17], both tumor and germline DNA were extracted using the Promega Maxwell 16 MDx DNA Purification Kits (Promega, Madison, WI). Whole-exome sequencing (WES) was performed using the Illumina HiSeq2500 (2×100bp) (Illumina, San Diego, CA). Reads were aligned to the GRCh37/hg19 reference and processed according to the Whole Exome Sequencing Test for Cancer - ExaCT1 pipeline v0.9 [16–18]. Total RNA was extracted for RNAseq using Promega Maxwell 16 MDx instrument. RNA integrity was verified using the Agilent Bioanalyzer 2100 (Agilent Technologies) and prepared for whole transcriptome analysis (RNA-Seq) in accordance with the standard Illumina mRNA sample preparation protocol (Illumina, San Diego, CA). Paired-end RNA-Seq at read lengths of 50 base pairs was performed with the HiSeq 2000 (Illumina). We performed sequence alignment using STAR_2.4 with human genome sequence build hg19, downloaded via the UCSC genome browser (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/).
Immune cell deconvolution and TCGA cohort validation
We utilized xCell, a gene signature-based algorithm, for the deconvolution of bulk RNA-sequencing data to interrogate the tumor immune landscape [19]. We evaluated transcriptome expression data for 64 immune and stromal cell types and the results were associated with WES results. Consensus clustering was performing using ConsensusClusterPlus R package. To validate our findings, the TCGA colon cancer cohort [6] (n = 477) was also interrogated, and the results were correlated with Consensus Molecular Subtypes of CRC (https://portal.gdc.cancer.gov).
Immunohistochemistry (IHC)
Cases were re-reviewed by a certified pathologist with subspecialty training in gastrointestinal pathology. Histologic features including tumor architecture, tumor-stroma interface, immune infiltration, and stromal response were recorded for each case. On a subset of tumors IHC was performed on sections of formalin-fixed paraffin-embedded (FFPE) tumor tissue using a Bond III automated immunostainer and the Bond Polymer Refine detection system (Leica Microsystems, IL, USA). The following antibodies and conditions (dilution, antigen retrieval solution and pH, antigen retrieval time) were used: CD86 (CellSignaling Technology E2G8P Rabbit mAb, 1:100, Sodium Citrate buffer, pH6, 30 min) and CD80 (MyBioSource MBS4380739 Mouse mAb, 1:100, Sodium Citrate buffer, pH6, 30 min).
Imaging Mass Cytometry
Per the manufacturer’s protocol, antibodies were conjugated in BSA and Azide-free format using the MaxPar X8 multimetal labeling kit (Standard BioTool). The antibodies were tested on control tissues (e.g., lymph node, tonsil) to validate the staining pattern as verified by study pathologists. Fifteen tumors (2 primary and 13 metastases) from 13 patients underwent in situ immune evaluation by imaging mass cytometry (IMC). The tissue samples were stained according to the manufacturer protocol following traditional immunohistochemical methods. Briefly, after 2 hours of warming the slides, these were deparaffinized in Citrisolv twice for 15 mins each followed by ethanol rehydration. Antigen retrieval was performed in a water bath for 30 minutes at pH 9.0. The slides were then washed and blocked using ThermoFischer SuperBlock solution for an hour. The slides were incubated overnight in antibody cocktail and washed the next day in TBS buffer and water. Lastly, DNA intercalator staining was performed by incubating the slides in 1:400 Ir solution for 30 mins at RT. The slides were washed and air-dried, ready to be scanned. Two regions of interest (ROI) were chosen per tumor, including tumor/normal interface when present.
Imaging Mass Cytometry Data Analysis
The IMC^™^ data was processed using the imc (v.0.1.4) package, available at [https://github.com/ElementoLab/imc] as we previously reported [20,21]. The 29-channel image was reduced into a probability map of nuclei, cytoplasm, and background. Cells were segmented by applying DeepCell (v.0.2.1) to the probability map. To identify the expression profiles in the multiplexed images, each channel of every cell was mean-aggregated. The Scanpy library was utilized for downstream preprocessing. A log transformation and z-score normalization truncated at 3 standard deviations was performed to bring all channels to the same scale. This was followed by batch correction using Harmony (v.0.0.9) [10.5281/zenodo.7351719] to mitigate sample-specific batch effects. To identify cell phenotypes, leiden clustering was applied with a resolution of 1.0[22]. Each resulting cluster was manually labeled using the matrixplot provided. For statistical analysis, the cellular density per ROI was calculated and a two-sided Mann-Whitney U-test was conducted, followed by a Benjamini-Hochberg multiple hypothesis correction to account for the repeated comparisons across cell types.
Results
Clinico-pathologic characteristics are representative of conventional colorectal carcinoma
Forty-one (41) colorectal carcinoma specimens from 31 unique patients (M:F ratio = 21:10, average age 55 years) were included in the study cohort (Table 1). Most (58%) were left-sided colorectal carcinomas. At the time of tissue acquisition, all but 3 patients had Stage IV disease, and most (77%) patients had previously received first-line chemotherapy prior to tissue collection. Nine samples (9) were from the primary tumor and 32 samples were taken from metastatic sites. The molecular characteristics of this cohort were fairly representative of conventional colorectal carcinoma, with frequent alterations in KRAS (74%), TP53 (71%), and APC (51%); The majority of tumors (95%) were microsatellite stable. Consensus molecular subtypes 2 and 4 were most common (27% and 24%, respectively). The follow up interval from date of diagnosis to last contact was 56 months (range 4–168 mo). Twenty-two (70%) patients were alive, and the mean survival for the cohort was 40 mo (range 21–70 months).
Colorectal Cancer is characterized by two clusters with distinct immune environments
Gene set enrichment analysis followed by unsupervised clustering revealed two distinct clusters, which varied in their immune composition (Fig. 1). One cluster, comprised of 21 samples (18 patients), contained relatively high numbers of activated dendritic cells and Th1 cells (activated immune microenvironment), whereas a second cluster, which included 20 samples (14 patients), contained increased inactive dendritic cells and basophils (inactivated immune microenvironment). Correlating with the above findings, dendritic costimulatory molecules and TH1 cell markers showed higher expression in the activated group (Supplementary Fig. 1).
The clinicopathologic features of the two subgroups are summarized in Supplementary Fig. 2. Patient demographics, tissue site (primary versus metastasis as well as liver vs non-liver metastasis), tumor grade and stage at diagnosis did not significantly differ between the groups, though right-sided tumors were more common in the suppressed immune group (p = 0.019). In addition, molecular analysis revealed more frequent KRAS mutations in the suppressed subgroup (75% versus 38%, p = 0.03) In keeping, CMS3 subtype was more common in the suppressed tumors while CMS2 subtype predominated in the activated group.
We then sought to confirm and further characterize these two immune subtypes by detailed histopathologic review and by Imaging Mass Cytometry. Fifteen samples (8 activated and 7 suppressed) underwent detailed histopathologic review. There was no difference in tumor architecture or histologic subtype, tumor budding, or necrosis. The presence of neutrophils, eosinophils, and plasma cells did not significantly differ between the two groups. However, tertiary lymphoid structures (TLS), by microscopic identification, were present in 5 tumors analyzed, 4 of which were categorized as activated. While these were numerous in two cases (both activated), most contained occasional TLS within the tumor periphery (Fig. 2a). In addition, a dense stromal response was present in 5 (71%) of the suppressed tumors whereas this response was only seen in 1 (14%) tumor within the activated group (p = 0.029) (Fig. 2b). Immunohistochemical staining for CD80 and CD86 (activated dendritic cells markers) was performed in representative cases, which showed strong positivity at the tumor stromal interface in activated tumors but not in suppressed tumors (Fig. 2c,d), supporting the RNA-Seq findings.
We focused the multiplexed IMC^™^ analysis on the immune composition of each tumor. To quantify the in situ expression of 26 protein markers, IMC^™^ was performed on a total of 83,898 cells from 15 tumors (11.14 mm^2^, 35 regions of interest). The antibodies included in the panel are listed in Supplementary Fig. 1and Fig. 3a. Qualitatively, IMC^™^ demonstrated a more robust T cell infiltrate throughout the peritumoral stroma within the activated group, with increased TLS, as was demonstrated on the hematoxylin & eosin-stained tissue slide. In suppressed tumors, the dense stromal response was highlighted, as well as an increased in macrophages (Fig. 2e,f). Quantitatively, CD8 T cells and macrophages were the most abundant inflammatory cells in the cohort (Fig. 3a, right panel). When comparing activated versus suppressed tumors, we found significant differences in the cell composition of the tumor microenvironment, with CD8 T cells being more frequent in the activated subgroup whereas macrophages were more often seen in the suppressed group, with increased M2/M1 ratio in the suppressed subgroup and increased expression of CD163 and CD206 (Fig. 3b,c). Dendritic cells were difficult to isolate through this methodology and the activity state could not be reliably determined, as signals for CD86 and CD80 by this methodology were weak and relatively nonspecific.
We next examined the immune microenvironment as it relates to tumor location or metastases site. Although the IMC analysis identified no significant difference in cell type proportion across primary or metastatic sites (Supplemental Table 2), we did observe that the immune activated v immunosuppressed clusters readily separate in both the primary tumor and in colorectal liver metastases clusters, whereas this distinction was less apparent with extrahepatic CRC metastases (Fig. 4). In patients with CRC liver metastases, we observe an inflammatory TIME enriched in CD8 and CD4 T cells and M1 macrophages, along with pro-tumor immune infiltrate of M2 macrophages, MDSCs and Tregs. The immune suppressed CRC liver metastasis cluster is also enriched in TGFb signaling and the immune checkpoint blockade signature 2 (Fig. 4), consistent with the observation that active liver metastases induce systemic immunosuppression and relative resistance to immunotherapy. Four patients had samples from multiple sites that underwent RNA-Seq analysis. Three patients had 2 samples each, whereas metastases from 5 distinct sites as well as the primary tumor were evaluated in one patient (Supplementary Table 3). In two patients, the metastatic tumors clustered similarly. However, in one patient the initial primary carcinoma was found to have a suppressed immune environment while the subsequent lung metastasis demonstrated an activated immune environment. Additionally, for the patient with 6 distinct tumor samples, all but one clustered as suppressed while the adrenal metastasis was classified as activated.
Distinct tumor immune microenvironments of colorectal carcinoma are associated with overall survival
To examine the activated and immune suppressed TIME CRC signature in a larger cohort, a similar gene set enrichment analysis with unsupervised clustering was performed on the publicly available RNA-Seq data from the TCGA primary colon carcinoma cohort, comprising 477 patients with newly diagnosed colorectal carcinoma. In contrast to our study cohort, these patients presented mostly with earlier stage disease and had not received therapy prior to sample acquisition. However, similar to the findings in our cohort, unsupervised clustering did separate the TCGA tumors into two separate groups, which varied predominantly based on their inactivated to activated dendritic cell ratio (Supplementary Fig. 2). There was no significant difference in stage of disease between the two groups, though we did again identify a significant association between KRAS mutational status and increased inactivated dendritic cells (p = 0.006). Finally, we looked at the survival probability in relation to the dendritic cell ratio. Those with increased inactive dendritic cells (i.e. the immune suppressed TME) had a lower overall survival probability compared with those who had more activated dendritic cells (i.e. activated TME) (Fig. 5, HR 1.66, p = 0.007).
Discussion
Herein, we identify two distinct clusters of advanced colorectal carcinomas with unique immune microenvironment signatures. Our findings suggest that this distinction is based predominantly on activation states of the immune cell composition, and largely independent of the CMS subtype or of driving oncogenic mutations. This study adds to the expanding literature which focuses on the importance of the tumor microenvironment in disease heterogeneity. Using currently available advanced technology, we demonstrate, similar to the findings by Galon et al. in this cancer subtype, that an activated immune environment (including CD8 T cells but also dendritic cells, tertiary lymphoid structures, among others) may be associated with improved patient prognosis. Further studies utilizing these techniques to better characterize the activation state of various immune cell subpopulations will help to uncover whether tumors with activated immune environments may be more sensitive to checkpoint inhibition, offering additional therapeutic implications.
The tumor microenvironment comprises a complex network of cell types such as lymphocytes, dendritic cells, myeloid cells, fibroblasts, endothelial cells and extracellular components (cytokines, chemokines, etc.), all of which heavily influence the ability of a tumor to grow and disseminate [23–25]. The characteristics of this environment are thought to have significant prognostic and even therapeutic implications in numerous cancer subtypes including ovarian, colorectal, lung and breast, and several studies have shown that the inflammatory network in particular plays a pivotal role in the evolution of cancer [26–28]. However, it is not just the density and cell type but also a cell’s specific phenotype which remains imperative in directing the immune response to be either immunostimulatory or immunotolerant, a feature that is becoming more evident as our ability to characterize these components advances. Although we demonstrate that an activated CRC TIME is associated with improved survival, it is not clear at this time if this improved survival is due to improved benefit with cytotoxic therapy, or rather a feature of a less aggressive CRC phenotype.
Cancer cells have evolved multiple mechanisms to escape immune surveillance, such as defects in antigen presentation machinery, upregulation of negative regulatory pathways, and the recruitment of immunosuppressive cell populations which ultimately result in inefficient cytotoxic response. Our ability to characterize and define the TIME has significantly evolved. Earlier studies relying on manual inspection of glass slides and single surface marker analysis by immunohistochemistry were limited in their ability to assess the dynamic nature of the TIME. This lack of specificity in defining subpopulations of cells may account for some of the inconsistencies in the literature between cell type and prognosis. Due to the complexity of the immune microenvironment that we now recognize, computational algorithms using bulk transcriptome data have become a popular approach to better understanding this interaction between tumor and host. The CMS classification scheme, for example, was one of the first subclassification studies using RNA-Seq data in colorectal carcinomas to incorporate features of the immune microenvironment in addition to molecular alterations within the tumor, in order to derive subclassifications [6]. This is pertinent in colorectal cancer, as multiple studies have demonstrated a correlation between the presence of mature T cells and dendritic cells and a more favorable prognosis, the former being the basis for the Immunoscore [9,12,13,29,30]. Similarly, the presence of B-cell and T-cell aggregates, also known as tertiary lymphoid structures (TLS) in the TIME, is considered a favorable prognostic feature as they promote T cell activation and antitumor effects[8,31]. Dendritic cells also play a prominent role in tumor response, as they vary significantly in their immunostimulatory or immunosuppressive activity at different stages of cancer progression and are incredibly dependent on local signaling [32,33]. In general, immature dendritic cells are peripherally located and non-mobile but are highly phagocytic and can be stimulated by various factors (including tumor antigens) to undergo maturation and activation, which then elicits an immune response by presenting antigens to T cells, inducing cytotoxic T lymphocyte immune response [34]. They also secrete chemokines that cause NK cells and T cell migration into the tumor and cytokines that maintain cytotoxic functions [35,36]. Not surprisingly, studies in a variety of solid tumors have found that increased dendritic cells, particularly those in the mature state with higher expression of costimulatory molecules including CD40, CD83, and CD86, are associated with improved prognosis [37–40]. On the other hand, tumor-associated dendritic cells have been found to often exhibit impaired function, including decreased uptake and presentation of antigens, reduced expression of costimulatory surface molecules, and inefficient migration, all of which may in reduced immune response and tumor evasion [41]. Studies have shown that tumor cells can release cytokines such as IL-6, IL-10 and TGF-B that cause dendritic cells to remain in an immature or immunosuppressive stage [33,42–44]. There are also metabolic inhibitors including IDO and arginase, which are produced by tumor associated macrophages (particularly the M2 phenotype) and upregulation of receptors such as CTLA4 and PD-1, all of which may be contributing to tumor immune escape [45,46].
The findings of the current study are unique in that we were able to evaluate a specific cohort of patients with advanced colorectal carcinoma, mostly Stage IV, who had already failed first line therapy. Within this cohort, we first identified two distinct groups through unsupervised clustering that differed predominantly in the activation state of the dendritic cells and other immune cells. Our findings are in keeping with other studies which have found an association between increased Th1 and activated dendritic cells and improved prognosis, whereas increased inactivated dendritic cells are generally associated with progression of disease [7,11,47]. Prior research has also characterized KRAS mutant CRC as having limited cytotoxic T cell infiltration, reduced T helper 1 responses, and reduced INF-gamma signaling, generating an overall immunosuppressive tumor microenvironment phenotype, which correlates with our finding of increased KRAS mutations in the subgroup with a suppressed TIME [48,49]. Another unique finding of the current study is that the reactivity state of the TIME did not depend on site of disease, either primary versus metastasis or location of metastasis. This is an important point, as prior studies have suggested that liver metastases are associated with immunosuppression, and patients with liver metastases have been shown to have minimal response to systemic immunotherapy [50]. However, we identified metastatic liver tumors with both activated and suppressed TIME, suggesting that a subset of patients with hepatic involvement may benefit from immunotherapy.
There are several recognized limitations of the current study. The cohort of patients with advanced colorectal carcinoma in which sufficient material available for RNA-Seq was relatively small. In addition, while review of the histomorphology and IMC^™^ analysis were performed in order to confirm our findings in situ, this comparison was somewhat limited by the fact that the FFPE tissue areas used for histopathology, IHC and IMC^™^ analysis may not exactly correspond to matched frozen tissue areas used for bulk RNA-Seq. Second, the IMC^™^ analysis was limited to discrete regions of interest that may not be representative of the entire tissue sample. Lastly, we were unable to fully characterize the immune population through IMC^™^ given the lack of additional immune markers in the available panel. Additional studies that can isolate dendritic cells and activations states are warranted.
In conclusion, this study supports the pertinent role of the immune microenvironment in tumor progression and patient prognosis, even within a group of patients with advanced stage disease who have failed multiple lines of therapy. We characterized the immune cell composition utilizing novel, more advanced technologies such as RNA-Seq and IMC^™^ in order to better understand the complexity and vital role that activity states play in determining response to tumor infiltration, thereby affecting response to therapy and overall patient outcome.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, Colorectal cancer statistics, 2017. CA Cancer J Clin 2017;67:177–93. 10.3322/caac.21395.28248415 · doi ↗ · pubmed ↗
- 2Shibutani M, Maeda K, Nagahara H, Fukuoka T, Iseki Y, Matsutani S, Tumor-infiltrating Lymphocytes Predict the Chemotherapeutic Outcomes in Patients with Stage IV Colorectal Cancer. In Vivo 2018;32:151–8. 10.21873/invivo.11218.29275313 PMC 5892643 · doi ↗ · pubmed ↗
- 3Ryuk JP, Choi G-S, Park JS, Kim HJ, Park SY, Yoon GS, Predictive factors and the prognosis of recurrence of colorectal cancer within 2 years after curative resection. Ann Surg Treat Res 2014;86:143–51. 10.4174/astr.2014.86.3.143.24761423 PMC 3994626 · doi ↗ · pubmed ↗
- 4Lieberman DA, Rex DK, Winawer SJ, Giardiello FM, Johnson DA, Levin TR. Guidelines for colonoscopy surveillance after screening and polypectomy: a consensus update by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2012;143:844–57. 10.1053/j.gastro.2012.06.001.22763141 · doi ↗ · pubmed ↗
- 5Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21:1350–6. 10.1038/nm.3967.26457759 PMC 4636487 · doi ↗ · pubmed ↗
- 6Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–7. 10.1038/nature 11252.22810696 PMC 3401966 · doi ↗ · pubmed ↗
- 7Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006;313:1960–4. 10.1126/science.1129139.17008531 · doi ↗ · pubmed ↗
- 8Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012;12:298–306. 10.1038/nrc 3245.22419253 · doi ↗ · pubmed ↗