Bactericidal human monoclonal antibody 1B1 shows specificity for meningococcal factor H binding protein variant 2 and displaces human factor H
Daniele Veggi, Chelsy C. Chesterman, Laura Santini, Ying Huang, Nicola Pacchiani, Jeannette Sierra, Lynn Chen, Jason Laliberte, Federica Bianchi, Roberta Cozzi, Elisabetta Frigimelica, Domenico Maione, Oretta Finco, Matthew J. Bottomley

TL;DR
A human antibody called 1B1 specifically targets a less-studied variant of a protein on meningococcal bacteria, helping to kill the bacteria and potentially improving vaccine design.
Contribution
The first detailed structural and functional analysis of an antibody with specificity for fHbp variant 2.
Findings
The crystal structure of the 1B1-fHbp variant 2 complex was determined at 2.4 Å resolution.
Key hotspots in the antibody-protein interface were identified through mutagenesis and binding studies.
1B1 displaces human factor H from fHbp variant 2, contributing to its bactericidal activity.
Abstract
Thousands of disease cases and hundreds of deaths occur globally each year due to invasive meningococcal disease. Neisseria meningitidis serogroup B (MenB) is the leading cause of such disease in developed countries. Two vaccines, 4CMenB and MenB‐fHbp, that protect against MenB are available and include one or two forms respectively of factor H binding protein (fHbp), a key protective antigen. Studies of circulating meningococci have identified over 1380 different fHbp amino acid sequences, which form three immunologically distinct clusters, termed variants 1, 2, and 3. Neither of the current vaccines contains a variant 2 antigen, which is less well characterized than fHbp variants 1 and 3. We characterized the interaction of fHbp variant 2 with humAb 1B1 using biochemical methods and live meningococcal assays. Further, we determined the crystal structure of the complex at 2.4 Å…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
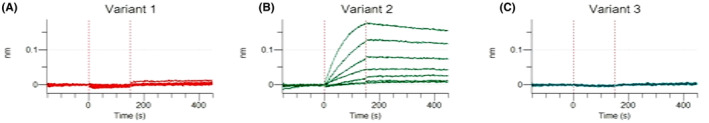 FIGURE 1
FIGURE 1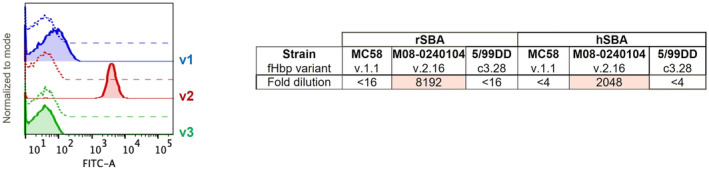 FIGURE 2
FIGURE 2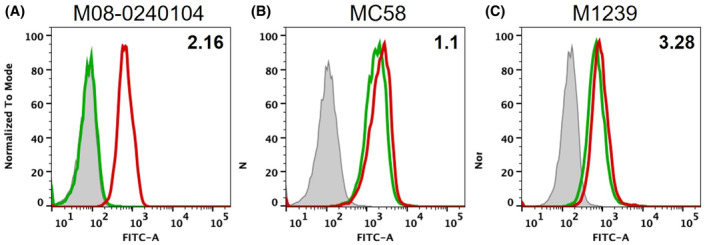 FIGURE 3
FIGURE 3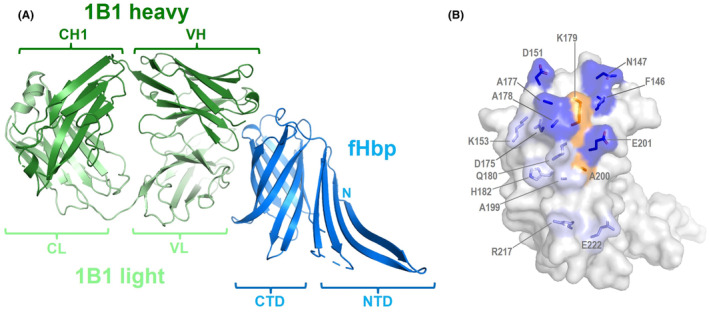 FIGURE 4
FIGURE 4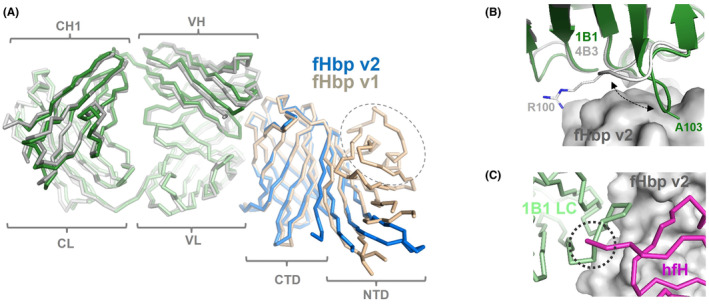 FIGURE 5
FIGURE 5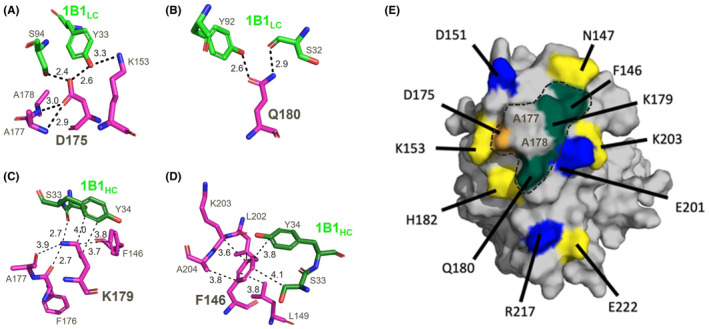 FIGURE 6
FIGURE 6| rSBA | hSBA | |||||
|---|---|---|---|---|---|---|
| MenB strain | MC58 | M08‐0240104 | 5/99DD | MC58 | M08‐0240104 | 5/99DD |
| fHbp variant | 1.1 | 2.16 | 3.28 | 1.1 | 2.16 | 3.28 |
| Titer | <16 | 8192 | <16 | <4 | 2048 | <4 |
| Wavelength | 0.966 |
| Resolution range | 49.66–2.41 (2.49–2.41) |
| Space group | P1211 |
| Unit cell | 77.15135.64121.53 90105.18 90 |
| Total reflections | 321,786 (33058) |
| Unique reflections | 92,813 (9128) |
| Multiplicity | 3.5 (3.6) |
| Completeness (%) | 99.7 (99.9) |
| I/sigma (I) | 10.0 (1.1) |
| CC1/2 | 0.997 (0.494) |
| Wilson B‐factor | 0.0656 (1.082) |
| R‐merge | 0.066 (1.17) |
| R‐meas | 0.078 (1.38) |
| Reflections used in refinement | 92,645 (9179) |
| Reflections used for R‐free | 4732 (453) |
| R‐work | 0.2050 (0.3568) |
| R‐free | 0.2693 (0.4282) |
| Number of non‐hydrogen atoms | 13,791 |
| Macromolecules | 13,495 |
| Ligands | 194 |
| Protein residues | 1798 |
| RMS (bonds) | 0.008 |
| RMS (angles) | 0.99 |
| Ramachandran favored (%) | 94.78 |
| Ramachandran allowed (%) | 4.65 |
| Ramachandran outliers (%) | 0.57 |
| Rotamer outliers (%) | 2.06 |
| Clashscore | 18.74 |
| Average B‐factor | 65.97 |
| Macromolecules | 66.14 |
| Ligands | 64.74 |
| Solvent | 55.72 |
| fHbp var 2 mutant | 1B1 affinity | 4B3 affinity | ||
|---|---|---|---|---|
| KD (nM) | Fold change | KD (nM) | Fold change | |
|
| 0.24 ± 0.03 | n/a | 0.002 | n/a |
| N147A | 0.26 ± 0.03 | 1.1 | 0.001 | 0.5 |
| H182A | 0.38 ± 0.11 | 1.6 | 0.002 | 1.0 |
| K153A | 0.45 ± 0.10 | 1.9 | 0.023 ± 0.04 | 11.5 |
| E222A | 0.50 ± 0.11 | 2.1 | 0.001 | 0.5 |
| R217A | 0.96 ± 0.15 | 4.0 | 0.001 | 0.5 |
| D151A | 1.13 ± 0.20 | 5 | 0.001 | 0.5 |
| E201A | 1.71 ± 0.44 | 7 | 0.042 ± 0.04 | 21 |
| F146A | 2.36 ± 0.25 | 10 | 0.739 ± 0.06 | 370 |
| K179A | 3.39 ± 0.47 | 14 | 4.8 ± 0.11 | 2390 |
| Q180A | 4.13 ± 0.75 | 17 | 1.35 ± 0.05 | 677 |
| D175A | 49.09 ± 32.35 | 205 | 0.002 | 1.0 |
| fHbp mutant | KD (nM) |
|---|---|
|
| No binding |
|
| No binding |
|
| 14.8 ± 3.1 |
|
| 12.1 ± 1.5 |
|
| 269 ± 150 |
|
| 32.2 ± 3.8 |
|
| 14.2 ± 1.4 |
| humAb name | Heavy chain | Light chain | |||||||
|---|---|---|---|---|---|---|---|---|---|
| V | D | J | IgCH | % identity VH V‐gene GL | V | J | IgCL | % identity VL V‐gene GL | |
| 1B1 | IGHV3‐48*04 | IGHD6‐13*01 | IGHJ4*02 | IgG1 | 98.98 | IGKV3‐20*01 | IGKJ1*01 | IgK | 96.90 |
| 4B3 | IGHV3‐11*01 | IGHD3‐16*01 | IGHJ4*02 | IgG1 | 88.10 | IGKV3‐20*01 | IGKJ2*01 | IgK | 90.24 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial Infections and Vaccines · Pneumonia and Respiratory Infections · RNA and protein synthesis mechanisms
INTRODUCTION
1
Thousands of disease cases occur annually due to Neisseria meningitidis infections.1, 2 Invasive meningococcal disease is highly feared because it can rapidly be fatal or cause severe long‐term sequelae such as limb amputation, hearing, and visual impairment, cognitive dysfunction, seizure, and behavioral disorders.3, 4 For over a decade, licensed glycoconjugate vaccines have effectively protected against meningococcal serogroups A, C, W135, and Y.5, 6 More recently, protection against meningococcal serogroup B (MenB) became possible through the development and licensure of two different recombinant protein vaccines (4CMenB and MenB‐fHbp).7, 8, 9 Real‐world data reviewed after millions of 4CMenB vaccinations in eligible infants in England showed high safety and effectiveness in preventing MenB disease.10, 11, 12
The two licensed MenB vaccines both contain factor H binding protein (fHbp), a key immunogen eliciting antibodies that exert complement‐mediated bactericidal activity.7, 13 Meningococcal fHbp is a 28 kDa surface‐exposed lipoprotein present on all known strains of N. meningitidis; its 3D structure comprises two β‐barrel domains connected by a short linker.14, 15, 16, 17 During meningococcal infections, fHbp binds the human complement regulatory factor H (hfH) thereby coating the bacterial surface, inhibiting activation of the complement alternative pathway and enabling evasion of host innate immunity.18, 19 Hence, the meningococcal virulence factor fHbp is an important target of vaccine‐elicited protective antibodies.
Over 1380 different amino acid sequence variants of fHbp have been reported to date,20 and they cluster phylogenetically as three variants.21 The three variants are immunologically distinct, that is one fHbp variant typically induces antibodies with high bactericidal activity against strains expressing similar variants (usually sharing ≥80% fHbp sequence identity), but with minimal activity against strains expressing different fHbp variants with lower sequence identity. The notable sequence variability of fHbp led vaccine developers to include multiple antigens in the MenB formulations: 4CMenB contains four different components (hence 4C) including fHbp variant 1, while MenB‐fHbp contains two components: fHbp variants 1 and 3. Neither vaccine contains fHbp variant 2, suggesting the possibility that vaccine efficacy might be improved by inclusion of a variant 2 antigen. To potentiate the design and development of such a vaccine, we sought to deepen our understanding of fHbp variant 2 structure and antigenicity.
Since 2018, crystal structures have been reported for fHbp variant 1 in complex with cross‐reactive human monoclonal antibodies (human mAbs, or humAbs) 1A1222 and 4B3,23 and for fHbp variant 3 in complex with humAb 1E6,24 collectively revealing the specificity and mechanism of action of these humAbs. However, to date, there are no reported structures of any mAbs bound to fHbp variant 2, and our structural understanding of wild type fHbp variant 2 is limited to its C‐terminal β‐barrel domain only, determined previously by x‐ray crystallography.25 The structure of an unnatural mutated form of fHbp variant 2 with increased thermostability was also determined previously.26 It is likely that the unusually low thermostability of the N‐terminal domain (NTD) of wild type fHbp variant 225, 27 hampered prior efforts to determine its structure. Consequently, in addition to lacking a wild type variant 2 structure encompassing its NTD, there are also currently no well‐defined epitopes specific to variant 2. Such knowledge gaps should be filled since both antigen structure and antigen‐specific mAbs can provide crucial information and act as key reagents during vaccine development.28, 29, 30
We previously reported a study of samples collected from three healthy adult 4CMenB vaccinees, in which we identified and characterized a total of 110 different humAbs (where each humAb was obtained from a single individual). Therein, we principally focused on a remarkable cross‐reactive humAb 1E6.24 However, that study also identified humAb 1B1, and in a qualitative binding assay using recombinant Fab produced in E. coli, 1B1 appeared to show specificity for fHbp variant 2 (see Figure 1 in Bianchi et al.24). To date, there are no other reports of a humAb with specificity for fHbp variant 2, and only one other antibody (JAR11, from mouse) with variant 2 specificity has been reported, for which little epitope mapping information was provided.31, 32 While it was unexpected to obtain from 4CMenB vaccinees a mAb with specificity for fHbp variant 2, it is readily conceivable that the donor from whom we identified mAb 1B1 might have been previously infected with a meningococcal strain expressing fHbp variant 2, thus giving rise to the corresponding B cell. Here, we report the first detailed characterization of the bactericidal humAb 1B1 and its structure in complex with wild type fHbp variant 2. We used a variety of biochemical and live meningococcal assays. Coupled with x‐ray crystallographic and structure‐guided mutagenesis studies, we provide insights into the structure of wild type fHbp variant 2, its recognition by humAb 1B1, and the molecular basis underlying the distinct variant 2 specificity observed.
MATERIALS AND METHODS
2
Molecular cloning, protein expression, and purification of wild type proteins
2.1
All wild type fHbp proteins were cloned, expressed and purified as described previously. The human antibody 1B1 was cloned, expressed and purified in both Fab and full‐length IgG1 formats, as described previously.24
Human samples
2.2
As described previously,24 the human samples from which 1B1 and 4B3 antibody sequences were derived were collected from three healthy adults immunized with the 4CmenB vaccine, in a clinical trial conducted in Krakow (Poland), approved by the Bioethics Committee of the District Medical Doctors Chamber in Krakow and conducted in accordance with the Declaration of Helsinki. The use of samples was performed upon written informed consent obtained from participants before the study‐specific procedures.
Site‐directed mutagenesis of fHbp proteins
2.3
Oligonucleotide primer pairs designed in‐house and synthesized externally (IDT) were used for site‐directed mutagenesis PCR reactions (NEB) to introduce mutations of interest into the appropriate template plasmid encoding fHbp variant of interest (pET‐21b‐v1.1, pET‐21b‐v2.16, or pET‐21b‐v3.28). Single colonies were picked, cultured, and plasmid DNA extracted for sequence confirmation.
Production and characterization of fHbp mutants for BLI binding studies
2.4
Plasmids encoding fHbp mutants were transformed into BL21 (DE3) Star cells (Novagen) according to the manufacturer's protocol. Three colonies were transferred into 500 μL of TB media in 24‐deep well blocks. Cultures were grown for 2–4 h at 37°C with shaking at 250 rpm. Cultures were diluted with 3.5 mL of TB media and transferred to 25°C for 30 min. 1 mM IPTG was added before overnight incubation of the cultures. Harvested cell pellets were re‐suspended in 750 μL of BugBuster MasterMix (Sigma). Protein was purified through parallel one‐step nickel affinity His‐tag purification on a Kingfisher instrument (ThermoFisher) using MagHis particles (Promega). Protein was bound to 100 μL of suspended MagHis particles, washed three times in 25 mM Tris pH 8.0, 300 mM NaCl, 20 mM Imidazole, and eluted in 25 mM Tris pH 8.0, 300 mM NaCl, 250 mM Imidazole. Protein concentration was determined by biolayer interferometry (BLI) using Anti‐His (HIS2, Fortebio) biosensors operated at 30°C in an Octet 384‐Red instrument (Fortebio). Binding to purified fHbp was used to establish a standard curve for the quantitation experiments.
Biolayer interferometry binding studies of fHbp/mAb interactions
2.5
Binding affinity of fHbp variants 1.1, 2.16, 3.28, and mutants thereof, to mAb 1B1 or 4B3 was determined with an Octet instrument using Anti‐Fc Human (AHC, Fortebio) biosensors at 30°C. Wild type and mutant fHbp proteins were diluted in Octet buffer (1 × PBS, 1% (w/v) BSA) with protein concentrations ranging from 150 to 1 nM. Octet biosensors were first dipped in a solution of Octet buffer to establish the initial baseline and then mAbs were coated onto the biosensors by submersion in a 10 mcg/mL solution of antibody. Sensors were re‐equilibrated in Octet buffer before transfer of the tips to the fHbp serial dilution. The association binding curve was measured by the increase in Octet BLI signal over time, followed by a dissociation phase with the sensors transferred back to Octet buffer. Curves were manually inspected for quality and binding curves that contain only noise were removed, leaving a minimum of five curves for fitting. Binding curves for association and dissociation were fit with the ForteBio Analysis 11.0 software to a 1:1 binding ratio in order to determine the equilibrium dissociation constant (K_D_) of the interaction. K_D_ values reported are the average and standard deviation of three independent measurements.
Flow cytometry
2.6
The ability of 1B1 mAb to bind antigen exposed on the surface of N. meningitidis bacteria was determined using a FACScan flow cytometer (FACSCanto II, BD Biosciences, San Jose, CA, USA). Bacteria grown until early‐log phase (OD_600_ of ~0.25) were incubated with monoclonal antibody at the concentration of 10ug/mL. Antibody binding was detected using an Anti‐Human IgG (H + L)‐FITC conjugated produced in goat (Invitrogen 31,529) at a 1:100 dilution. Bacteria plus PBS‐1%BSA and secondary antibody were used as negative control. A total of 10,000 events for each sample was collected with the cytometer and FlowJo™ Software v.10 Flow Jo was used to analyze the data.
Inhibition of binding of fH
2.7
The ability of the HumAb to inhibit binding of fH to live bacteria was measured by flow cytometry. Bacterial cells grown until mid‐log phase (OD600 of ~0.5) were incubated with anti‐fHbp mAb (50 μg/mL in PBS‐1%BSA buffer) for 30 min at room temperature, followed by the addition of purified human fH (50 μg/mL for MC58 and M1239, 10 μg/mL for UK104), which was incubated for an additional 30 min at room temperature in a final reaction volume of 100 μL. fH binding was detected with a goat polyclonal antiserum to fH (Calbiochem 341,276) diluted 1:100 and incubated for 30 min at room temperature, followed by additional 30 min incubation with a donkey anti‐goat IgG–fluoresceinisothiocyanate (FITC) conjugate (Jackson Immunoresearch 705.095.003) diluted 1:100 in PBS‐1%BSA buffer.
Serum bactericidal assay
2.8
Bacteria grown until early log phase (OD_600_ of ~0.25) were diluted in Dulbecco's Phosphate Buffered Saline (DPBS) containing 1% bovine serum albumin (BSA) and 0.1% glucose at the working dilution of 10^4^–10^5^ and incubated with serial two fold dilutions of test monoclonal antibody starting from a concentration of 125 μg/mL. Serum bactericidal titers were defined as the monoclonal antibody dilution resulting in 50% decrease in CFU per milliliter after a 60‐min incubation of bacteria with the reaction mixture compared to the control CFU per milliliter at time zero. Pooled baby rabbit sera (Cedarlane, Burlington, Canada) or human serum obtained from volunteer donors under informed consent, were used as a complement source for rSBA or hSBA respectively.
Protein crystallization, diffraction data collection, and processing
2.9
To form the fHbp/Fab complex, 7 mg of fHbp variant 2.16 and 5 mg of Fab 1B1 were co‐incubated overnight at 4°C and further purified by size exclusion chromatography to remove excess protein using a prepacked HiLoad 26/60 Column Superdex 75 Preparation Grade (GE Healthcare). The fHbp/Fab complex at a concentration of 15 mg/mL in buffer containing 50 mM Tris–HCl pH 8.0 and 200 mM NaCl were screened in over 500 different crystallization conditions using prepacked 96 deep‐well blocks commercialized by Molecular Dimensions (Newmarket, UK) using a Crystal Gryphon robot (Art Robbins Instruments, Sunnyvale, CA, USA). After incubation at 21°C using a RockImager‐182 system (Formulatrix Inc.), crystals were obtained using a reservoir solution containing 0.09 M sodium Nitrate, Sodium Phosphate dibasic, Ammonium Sulphate, 0.1 M Buffer Imidazole, MES monohydrate, pH 6.5 and as precipitant mix 25% v/v MPD, 25% PEG 100, 25% w/v PEG 3350.
Crystals were soaked in the mother liquor supplemented with 15% ethylene glycol prior to cryo‐cooling in liquid nitrogen. Diffraction was tested at the ESRF, beamline ID30A‐1, and several full datasets were collected at 100 K, wavelength λ = 0.966 Å, on a Pilatus3_2M Detector (see Table 2). Diffraction datasets were indexed, integrated, and scaled with XDS33 and Aimless,34 via the CCP4 suite.35 Crystals of the fHbp‐1B1 complex belonged to space group P1211, with the asymmetric unit containing three complexes and having a solvent content of 52% (Matthews coefficient of 2.69 Å^3^/Da). The structure was solved by molecular replacement (MR) using Phaser software.36 The search model templates included three distinct structures. First, we used the fHbp variant 1 from a previously determined 2.0 Å‐resolution structure (pdb 3KVD).15 Prior to MR, the fHbp template was modified by mutation to alanine residues at several positions where fHbp variants 1 and 2 differ in sequence. Second, we included the variable region of the Fab 4B3 structure and, third, the constant region of the Fab 4B3 structure—where both chains were separately extracted from Fab 4B3 in the complex structure determined previously (pdb 6XZW).23 Again, prior to MR, the Fab‐derived search models were alanine‐mutated or trimmed in the more variable regions. Thus, three separate MR search models were used (rather than the entire structure of the previous fHbp/4B3 complex) in order to minimize risk of bias in the MR solution.
Structure refinement
2.10
Initial MR solutions were subjected to cycles of manual building in Coot37 and subsequently refinement with Phenix.refine.38 Structure figures were created with Pymol (www.pymol.org). Structure factors and atomic coordinates have been deposited in the Protein Data Bank (www.wwpdb.org)39 with accession code PDB 8BK2.
RESULTS
3
HumAb 1B1 shows high affinity and distinct specificity for fHbp variant 2
3.1
Therefore, here, we first sought to confirm the apparent specificity of humAb 1B1 in a quantitative binding analysis based on biolayer interferometry (BLI), using a full‐length IgG form of recombinant 1B1 produced in mammalian cells, which are generally considered a more appropriate expression system than E. coli for the faithful production of heterologous mammalian proteins (see Section 2).
Antibody–antigen interactions were measured for humAb 1B1 captured via its Fc region, with fHbp proteins provided in solution. HumAb 1B1 showed strong interactions with fHbp variant 2 (equilibrium binding dissociation constant, K_D_, 0.24 ± 0.03 nM), while no interactions with fHbp variants 1 or 3 were detected (Figure 1). (We tested the often‐used fHbp exemplars (variants 1.1, 2.16 and 3.28), it cannot easily be excluded that there may exist some mutated forms of variants 1 or 3 that exhibit some affinity for humAb 1B1). These data confirm that humAb 1B1 has distinct specificity for fHbp variant 2 and forms a stable complex with moderately high affinity.
Biolayer interferometry (BLI) experimental data exploring the interaction of humAb 1B1 with fHbp variant 1 (panel A), fHbp variant 2 (panel B) and fHbp variant 3 (panel C). First, BLI biosensors suitable for capturing the Fc region of human antibodies were dipped into a solution of humAb 1B1 in order to coat the biosensor surface with antibody and reach equilibrium. Then, at the start of the binding experiment (Time = 0 (s)), the biosensors coated with humAb 1B1 were transferred into seven fHbp solutions representing a serial dilution of 150–1 nM fHbp antigen, for 150 s. Upon binding of any macromolecule to humAb 1B1, the BLI biosensors register an increase in the light interference pattern (y‐axis shows this increase as wavelength shift, in nm), proportional to the amount of antigen bound. After 150 s, the biosensors are transferred into antigen‐free buffer solution, resulting in dissociation of antigen from biosensors and a decrease in signal over time. Hence, the notable BLI responses observed only in panel B indicated binding to humAb 1B1 only for fHbp variant 2, and not for fHbp variants 1 or 3.
HumAb 1B1 preferentially binds and mediates bactericidal activity against live meningococci expressing fHbp variant 2
3.2
Following the in vitro binding analysis, we examined the 1B1/fHbp interaction in a more physiological context, by monitoring binding of humAb 1B1 to the surface of live meningococci. Using flow cytometry, we found that humAb 1B1 bound extensively to MenB strain M08‐0240104 which expresses variant 2, but only at very low levels to strain MC58 (expresses variant 1) and not detectably to strain M1239 (expresses variant 3) (Figure 2).
Flow cytometry data—the histograms display the frequency distribution of the bacteria (y‐axis) versus fluorescence intensity (x‐axis). The FITC‐fluorescence detected on the surface of bacteria after the incubation with 10 μg/mL of mAb 1B1 was acquired in logarithmic scale (blue line, MC58 strain; red line, M08‐0240104 strain; green line, M1239 strain). For each MenB strain, dotted line histograms represent negative control bacteria incubated with PBS and anti‐human IgG FITC‐conjugated. Only for fHbp variant 2 the peak fluorescence notably shifted to the right (fluorescence measurement >103), indicating that mAb 1B1 binds significantly only to the meningococcal strain displaying fHbp variant 2.
We also examined whether the binding to bacteria matched the functional activity of humAb 1B1 in standard serum bactericidal activity (SBA) assays, the established surrogate of protection for N. meningitidis.40, 41 In SBA assays using either rabbit or human sources of complement, humAb 1B1 was highly effective at killing MenB strain M08‐0240104 (expresses variant 2) but was not effective at killing strains expressing variants 1 or 3 (Table 1). Overall, the observations from these in vivo experiments align with the in vitro binding study, demonstrating that humAb 1B1 shows distinct specificity for fHbp variant 2 in both biochemical and live meningococcal functional assays.
HumAb 1B1 competes with human factor H for binding to meningococci expressing fHbp variant 2
3.3
It is relatively unusual to identify an anti‐fHbp mAb that, in the absence of additional mAbs, is potent in the human complement SBA (hSBA) assay.23, 43 Indeed, to date, only three highly similar humAbs (4F9, 4B3, and 3G7) have alone shown high potency in the hSBA assay. Previously, we determined the structure of fHbp variant 1 bound to humAb 4B3, revealing that 4B3 bound to a site on fHbp that prevented the simultaneous binding of hfH, thus, at least in part, explaining the high potency of humAb 4B3.23 Therefore, we sought to understand whether humAb 1B1 could similarly compete with hfH for binding to fHbp on live meningococci. Using flow cytometry, we found that humAb 1B1 was indeed effective at preventing the binding of hfH to MenB strain M08‐0240104 (expresses variant 2) but had no detectable impact on the binding of hfH to MenB strains MC58 or M1239 that express fHbp variants 1.1 and 3.28, respectively (Figure 3). These findings suggest that humAb 1B1 binds specifically to a region of fHbp variant 2 that overlaps with the binding site of hfH and this competitive binding capability may underlie the high potency of 1B1 in SBA assays.
Flow cytometry data showing binding of human factor H (hfH) to live meningococci displayed as FITC‐fluorescence intensity in the x‐axis: the histograms represent the fluorescence of bacteria pre‐incubated with humAb 1B1 (green line) or with buffer only (red line). Gray‐filled areas represent negative control bacteria incubated without humAb 1B1 and hfH, only with buffer and secondary FITC‐conjugated antibodies. Panel A: for v2.16, hfH binding was detectable when the bacteria were pre‐incubated with buffer alone (red histogram) whereas pre‐incubation with buffer containing humAb 1B1 inhibited the hfH binding (green histogram). In panels B and C (testing bacteria expressing variant 1.1 or 3.28), the equal florescence intensity of green and red histograms denotes that pre‐incubation with humAb had no effect on hfH binding.
Wild type fHbp variant 2 structure reveals similarities and differences with known fHbp structures
3.4
To deepen our understanding of its hitherto only partly characterized structure, we attempted to crystallize wild type fHbp variant 2. We failed to crystallize the protein alone, likely due to the known low thermostability of its N‐terminal domain (NTD; T_m_ ~ 42°C).25, 27 Consequently, we sought to promote crystallization by first forming the complex of wild type fHbp variant 2 with Fab 1B1 (the monovalent Fab form of humAb 1B1), the structure of which had not been previously determined.Strongly diffracting crystals of the complex were obtained, and the structure was solved by the MR technique and refined at 2.4 Angstrom (Å) resolution, with the R‐factor and free R‐factor converging at 20.5% and 26.9%, respectively (Table 2). The asymmetric unit contained three copies of the Fab‐antigen complex (hence, nine polypeptide chains in total) wherein all copies were essentially identical. The final refined models covered the entire Fab heavy and light chain sequences, and the fHbp variant 2 sequence from residue D76‐E261 (except loop residues 121–130, also previously unobserved in fHbp variant 1 structures, likely due to high flexibility of this loop23). Intriguingly, no electron density was observed for fHbp variant 2 residues up to N75, suggesting this N‐terminal region was disordered in the crystal. This new data expands our understanding of the wild type fHbp variant 2 structure, while the enigmatic N‐terminal region still remains partially undefined.
We compared our wild type fHbp variant 2 structure with previously reported fHbp structures. As expected, the structure showed moderate similarity with fHbp variant 3 (overall alpha‐carbon atom root mean square deviation [rmsd] 0.83 Å), high similarity with variant 1 (rmsd 0.67 Å), higher similarity with the unnatural double‐mutant stabilized form of variant 2 (rmsd 0.58 Å),26 and very high similarity in the available C‐terminal domain (CTD) sub‐region of wild type variant 2 (rmsd 0.38 Å).25 Only upon determination of our wild type fHbp variant 2 structure was it possible to observe differences in the NTD structure derived from previous genetic engineering efforts performed to create the double‐mutated stabilized variant 2 (pdb 4z3t).26 Specifically, the wild type NTD β‐strand spanning S133‐G141 makes fewer contacts to the neighboring CTD than the corresponding β‐strand R127‐G136 in the engineered stabilized mutant. This structural difference together with disorder in wild type NTD residues 1–75 may underlie the low thermostability of the wild type variant 2.
Co‐crystal structure reveals fHbp variant 2 epitope targeted by humAb 1B1
3.5
The co‐crystal structure of wild type fHbp variant 2 bound to Fab 1B1 revealed all the molecular determinants at the epitope/paratope interface. The overall structure of the complex shows Fab 1B1 projecting all six complementarity determining regions (CDRs) onto a convex surface‐exposed region of the fHbp CTD β‐barrel, while the fHbp NTD does not contribute to the interaction (Figure 4A). Thirteen fHbp CTD residues are directly involved in binding to Fab 1B1 via 14 hydrogen bonds and 2 salt‐bridges (Figure 4B), while 6 fHbp residues make van der Waals' interactions with the Fab. Altogether, the Fab covers a surface area on fHbp of 744 Å^2^, a typical size for an epitope.44, 45
Co‐crystal structure of Fab 1B1 bound to fHbp variant 2 reveals epitope detail. (A) Fab 1B1 heavy and light chains (dark green, light green, respectively) in contact with fHbp (blue). Cartoon labels show variable heavy and light chain regions (VH, VL), constant heavy and light chains (CH1, CL), and fHbp N‐terminal and C‐terminal domains (NTD, CTD). (B) Semi‐transparent surface of fHbp (gray) with the following color scheme: dark blue/light blue patches show fHbp residues (side chain sticks shown) used to bind Fab 1B1 heavy/light chain via H‐bonds and salt bridges; orange/yellow patches show residues making van der Waals' interactions with heavy/light chain. Compared to panel A, the image is enlarged and rotated ~90° around the vertical plane (Fab 1B1 is not shown in panel B).
It is notable that humAb 1B1 binds to fHbp variant 2 in a region highly similar to where the cross‐protective humAb 4B3 binds to fHbp variant 1, suggesting that this might be a particularly immunogenic region of fHbp (Figure 5A). Of the fifteen total variant 2 epitope residues identified (Figure 4B), 12 corresponding variant 1 residues are present in the 4B3 epitope. However, while the two Fabs approach fHbp with very similar orientations, their CDR H3 loops have very different conformations and engage some different epitope residues (Figure 5B).
Similarities and differences of Fabs 1B1 (variant 2‐specific) and 4B3 (cross‐reactive) in binding to fHbp. (A) Fab 1B1 (green) and Fab 4B3 (gray) share the same angle of approach to fHbp variant 1 (wheat) and variant 2 (blue). The dashed oval shows the small N‐terminal region of variant 1 which is unobserved in our electron density maps of variant 2, likely due to local disorder. (B) The tertiary structures of Fabs 1B1 and 4B3 are highly superimposable (rmsd 0.54 Å for the VH regions) but the CDR3 loops diverge in opposite directions (see black dashed arrows), enabling the Fabs to engage slightly different epitopes. By sequence alignment, A103 and R100 are corresponding residues, but differ in spatial organization by 4.3 Angstroms, highlighting their different CDRH3 conformations (C) Superposition of the fHbp/1B1 and fHbp/hfH structures (overall rmsd 1.6 Å) shows how the N‐terminal region of hfH (magenta) and the CDR L2 loop of 1B1 (pale green) would clash (dotted circle) upon binding fHbp variant 2 (gray surface), thus preventing simultaneous binding.
The structure also suggested the potential molecular basis for the high functional activity of humAb 1B1. In short, by in silico superposition of the fHbp structures of the variant 2/1B1 complex and the variant 1/hfH complex,17 we observed that 1B1 and hfH could not compatibly bind to fHbp at the same time, due to steric hindrance. In particular, the CDR L2 of the 1B1 light chain would clash with the N‐terminal region of hfH domain 6 (Figure 5C).
An epitope hotspot drives the antigen–antibody interaction
3.6
To better understand the antigen–antibody interaction, we performed an extensive alanine scanning study, in which all eleven non‐alanine fHbp epitope residues were individually mutated to alanine. Mutated fHbp proteins were purified and tested via BLI assay for binding to humAb 1B1. The different alanine mutations had varying effects on the strength of the interaction, ranging from essentially conferring no change, up to a 200‐fold decrease in binding affinity (Table 3, left). Four individual mutations resulted in a relatively large loss of affinity (K_D_ values increased ≥10‐fold), indicating important roles in binding for residues F146, K179, Q180, and especially D175. These key mutations likely abolished one or more H‐bonds and/or reduced van der Waals interactions with the 1B1 light chain CDRs L1 and L3 and the heavy chain CDR H1 (Figure 6). These mutational data suggest a hotspot in the “centre/upper‐right region” of the epitope (Figure 6E) as the key antigenic area driving the strength of the antigen–antibody interaction. Intriguingly, five of these eleven mutations also decreased the binding affinity of fHbp variant 2 to humAb 4B3 (Table 3, right), though with a different rank order of impact on binding strength. Notably, despite its proximity to the hotspot residues, the D175A mutation surprisingly had no impact on binding to humAb 4B3. These data demonstrate that while the two humAbs 1B1 and 4B3 appear to bind the same site on fHbp, they achieve high binding affinity in different ways.
Experimentally verified molecular determinants of the interaction between humAb 1B1 (green) and fHbp variant 2 (magenta). (A) fHbp D175 is central to a multi‐valent H‐bond network, with two H‐bonds to Y33 and S94 on the Fab 1B1 light chain (1B1LC); (B) fHbp Q180 makes two H‐bond contacts to S32 and Y92 on 1B1LC; (C) fHbp K179 forms one H‐bond with S33 and van der Waals (VdW) interactions with Y34 on 1B1 heavy chain (1B1HC); (D) fHbp F146 sits in a hydrophobic pocket on fHbp and makes VdW interactions with S33 and Y34 on 1B1HC; (E) Surface plot of fHbp variant 2 (gray) with epitope residues colored according to fold‐increase in KD when mutated to alanine (orange, 200×; teal, 10×; blue, 4×; yellow, <2×). The epitope hotspot lies within the dashed‐line circle; the interface with 1B1 includes A177 and A178 which make VdW interactions that support binding; these residues were obviously not amenable to the alanine scanning approach.
Molecular basis for fHbp variant 2 specificity of humAb 1B1
3.7
Interestingly, all four key epitope hotspot residues (F146, D175, K179, and Q180), are identically conserved in fHbp variants 1.1 and 3.28, suggesting that, while important for the strength of binding, they are unlikely to account for the specificity of humAb 1B1 for fHbp variant 2. Rather, the epitope peripheral residue D151, for which the alanine mutation caused approximately five‐fold lower binding affinity to humAb 1B1, is a notable epitope residue not at all conserved in both variant 1 and 3 (it is replaced by glycine, which has no side chain). Also, H182 is mutated to N and Y in variants 1 and 3, respectively, but since the H182A mutation had little effect on binding strength, this residue seemed less likely to be important for specificity. Overall, these observations suggested that the acidic side chain of D151 may provide the crucial interactions specific for binding to humAb 1B1 (namely, two H‐bonds to Ser56 and Ser58 in CDR H2). Further, given the shape complementarity of the epitope/paratope interface in the hotspot region involving Ala177 and Ala178 (Figure 6E), it was conceivable that their switch to the bulkier Thr and Lys residues in variant 3 might obstruct binding. To explore these hypotheses, we first made a series of mutations in fHbp variant 3, attempting to create gain‐of‐function mutants that could bind to humAb 1B1. Using the BLI assay to test humAb 1B1 interactions with fHbp variant 3 mutants, we found that single mutants G151D and T177A were still unable to bind 1B1, whereas the double mutant T177A/K178A and the triple mutant G151D/T177A/K178A exhibited quite tight binding (Table 4, upper), albeit ~50‐fold weaker than variant 2. These data suggest the main reason wild type fHbp variant 3 does not bind to humAb 1B1 is due to steric hindrance at position K178.
Since fHbp variant 1 contains Alanines like A177/A178 in variant 2, a different reason must underlie its inability to binding humAb 1B1. Indeed, compared to variant 2, while variant 3 replaces D151 simply with glycine, in variant 1 there is a Glu‐Gly dipeptide motif, in a conformation likely incompatible with forming bonds to 1B1. To explore these observations experimentally, we also tested fHbp variant 1 mutants, and found that moderate binding to humAb 1B1 could be achieved by the simultaneous E151D mutation and G152 deletion (Table 4, lower). Binding affinity was further increased by D201E and L217R mutations, which flank the epitope hotspot (Figure 4B). Collectively, these mutational studies of fHbp variants 1, 2 and 3 help to explain the variant 2 specificity of humAb 1B1.
humAbs 1B1 and 4B3 have different germline origins yet bind equivalent epitopes
3.8
The high degree of structural similarity exhibited by the 1B1/variant 2 and 4B3/variant 1 complexes (Figure 5A) prompted us to ask whether the two humAbs share similar sequences despite deriving from different donors. We analyzed the nucleotidic sequences of both humAbs using NCBI igblastn version 1.18.0.46 The VDJ gene sequences revealed that the heavy chains of humAbs 1B1 and 4B3 are both IgG1 isotype but have different genetic origins, while the light chains (IgK type) are more similar (Table 5). The 1B1 and 4B3 heavy chain variable regions (VH) share only 74% amino acid sequence identity, while their light chain variable regions (VL) share 83% sequence identity. Interestingly, while all six CDRs contact the antigen in both complexes, it is the heavy chain of 1B1 that makes the majority of contacts with the epitope hotspot on fHbp variant 2 (Figure 6E), thus further explaining the different binding capabilities of the two humAbs. Nucleotidic sequence alignments suggested that overall humAbs 1B1 and 4B3 heavy chains shared 99% and 88% sequence identity with their inferred V‐gene germline precursors (Table 5), highlighting diverse degrees of somatic hypermutation of these two humAbs, enabling their distinct specificities despite their remarkably similar structures, epitope regions, and angles of approach to the antigens.
DISCUSSION
4
N. meningitidis is a globally important pathogen causing invasive meningococcal disease, often with severe sequelae and fatal in 10%–20% of cases. Two highly effective serogroup B meningococcal vaccines are now available, containing fHbp variants 1 and 3 (MenB‐fHbp vaccine), or fHbp variant 1 and other antigens (4CMenB vaccine). Since the breadth of protection afforded by such vaccines might be further increased by the inclusion of additional, diverse fHbp antigens, we sought to characterize the immunologically distinct fHbp variant 2. The latter is relatively uncharacterized and our vision was that determination of its structure and antigenicity might enable the design and development of improved vaccines.
Since we, like others previously, were not able to crystallize the wild type fHbp variant 2 protein alone, we sought a high affinity variant 2‐specific mAb, the Fab form of which might “chaperone” crystallization. Such co‐crystallization methods have been successfully used previously.47 In an earlier study of numerous humAbs, we had preliminarily identified an apparent variant 2‐specific Fab, termed 1B1.24 In the present work, we performed a detailed study of 1B1, both in IgG1 and Fab formats. In doing so we have characterized, to the best of our knowledge, the first fHbp variant 2‐specific humAb. We have revealed at the molecular level the ability of 1B1 to compete the interaction of fHbp with human fH, thus increasing the susceptibility of the bacteria to complement‐mediated bacteriolysis and explaining its mechanism of action as a highly potent bactericidal antibody. Moreover, Fab 1B1 was indeed able to chaperone the crystallization of wild type fHbp variant 2, enabling structure determination at high resolution.
Analysis of the paratope/epitope interface formed between Fab 1B1 and fHbp variant 2 revealed the basis of its antigen specificity. A key molecular determinant of the interaction is an H‐bond between 1B1‐Ser56 and fHbp variant 2 Asp151, the latter being part of the 150‐PDGKA‐154 loop. A crucial local difference is the insertion of a second Gly in fHbp variant 1 (PEGGRA) which distorts the loop structure and likely prevents the variant 1 Glu residue from making the productive interaction with 1B1‐Ser56. This and other observations described above explain the antigen specificity of humAb 1B1 and were sufficient to inform the design of mutations in fHbp variants 1 and 3 that conferred gain‐of‐function to enable binding to humAb 1B1.
Unexpectedly, Fab 1B1 was found to bind fHbp in a manner extremely similar to that of the cross‐reactive Fab 4B3. Notwithstanding an almost identical angle of approach used by the two Fabs, and high structural similarity of their framework regions (Figure 5A), Fab 1B1 did not bind to wild type fHbp variants 1 or 3, while Fab 4B3 is cross‐reactive. Analyses of the sequences of these two humAbs suggests they have different origins, and yet converged upon a similar spatial solution to engaging this key antigen. Indeed, not only do they bind to similar epitopes, both Fabs can also prevent the binding of human fH to the antigen. This finding is particularly relevant, since the 1B1 structure hereby provides a second example (after 4B3) of a humAb able to bind the hfH binding site, confirming that this surface region on fHbp is at least partially or temporarily exposed and immunogenic in humans. The latter topic was previously debated, since it was thought that the region was masked from the immune system due to binding of human fH. The data we present here support the hypothesis that the site is accessible and immunogenic, perhaps because the circulating human fH concentration is simply insufficient to occupy all possible sites on fHbp and completely shield them from the immune system.
Despite having provided the first structural and functional insights into a human mAb with specificity for fHbp variant 2, there remain some unanswered questions. For example, despite Fab 1B1 having aided crystallization of fHbp variant 2, there was a notable absence of some N‐terminal fHbp residues (V9‐N75) in the crystallographic electron density maps. This prompts the question as to whether the N‐terminal region is naturally disordered or, rather, was destabilized by the presence of the mAb and/or the buffer conditions used for crystallization, or a combination of both factors. The former explanation seems more likely since several studies of fHbp variant 2 in solution have indicated a low thermal stability of its N‐terminal domain. It is unclear from an evolutionary viewpoint why the variant 2 protein should have maintained this low stability, which plausibly leads to its more rapid proteolytic turnover on the bacterial surface and consequent loss of function in situ. Further, our studies were limited to typical exemplars of the fHbp variants 1, 2 and 3 (namely, variants 1.1, 2.16, and 3.28), for which humAb 1B1 displayed distinct specificity for variant 2. Considering that there are over 1380 distinct fHbp amino sequences, it is possible that some sub‐variants of variants 1 or 3 may display modest affinity for humAb 1B1. Finally, it was unexpected to obtain a mAb with specificity to fHbp variant 2. We speculate that the individual donor from whom we identified mAb 1B1 might have been previously infected with a meningococcal strain expressing fHbp variant 2 and/or our in vitro assays had features that enabled selection of such a B cell. Further studies will be required to address such questions.
In summary, our structural and functional investigations have shed additional light on the nature of the human immune response to meningococcal fHbp antigens. Specifically, the generation of new data on the relatively uncharacterized variant 2 fHbp protein should enable the design of alternative novel candidate vaccine antigens, potentially by rational design to improve molecular stability while ensuring the presence of demonstrated protective epitopes capable of eliciting potent bactericidal antibodies. Moreover, variant 2‐specific antibodies may be useful for the characterization of this specific antigen in the context of multi‐antigen vaccines. Overall, the data presented may be of particular benefit to support the development of meningococcal vaccines, and more broadly demonstrate the value of combining human immunology and structural biology studies to deepen our understanding of pathology and vaccinology.
AUTHOR CONTRIBUTIONS
D.V., C.C.C., R.C., F.B., L.C., J.L., Y.H., A.B.C., M.J.B. designed and/or conducted the studies, including data collection and data analysis. D.V., C.C.C., L.S., and M.J.B. prepared the manuscript. All authors reviewed and approved the final manuscript. All authors had full access to the study data.
FUNDING INFORMATION
This study was performed at GSK Vaccines Srl (Siena, Italy) and at GSK Vaccines (Rockville, US), both locations are part of the GSK group of companies. FB was the recipient of a PhD fellowship at the University of Florence.
CONFLICT OF INTEREST STATEMENT
All authors were employees of the GSK group of companies at the time of the study, with the exception of FB, who held a PhD studenship at the University of Florence, Italy. The authors DV and MJB are named inventors on patent applications related to fHbp polypeptides. MJB, DM report ownership of GSK shares.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1ECDC . European Centre for Disease Prevention and Control. Invasive meningococcal disease. ECDC Ann Epidemiol Rep. 2017;27:1‐10.
- 2Rosenstein NE , Perkins BA , Stephens DS , Popovic T , Hughes JM . Meningococcal disease. N Engl J Med. 2001;344:1378‐1388.11333996 10.1056/NEJM 200105033441807 · doi ↗ · pubmed ↗
- 3Pace D , Pollard AJ . Meningococcal disease: clinical presentation and sequelae. Vaccine. 2012;30(Suppl. 2):B 3‐B 9.22607896 10.1016/j.vaccine.2011.12.062 · doi ↗ · pubmed ↗
- 4Olbrich KJ , Muller D , Schumacher S , Beck E , Meszaros K , Koerber F . Systematic review of invasive meningococcal disease: sequelae and quality of life impact on patients and their caregivers. Infect Dis Ther. 2018;7:421‐438.30267220 10.1007/s 40121-018-0213-2PMC 6249177 · doi ↗ · pubmed ↗
- 5Pace D , Pollard AJ , Messonier NE . Quadrivalent meningococcal conjugate vaccines. Vaccine. 2009;27(Suppl. 2):B 30‐B 41.19477560 10.1016/j.vaccine.2009.05.003 · doi ↗ · pubmed ↗
- 6Ruiz Garcia Y , Abitbol V , Pellegrini M , Bekkat‐Berkani R , Soumahoro L . A decade of fighting invasive meningococcal disease: a narrative review of clinical and real‐world experience with the Men ACWY‐CRM conjugate vaccine. Infect Dis Ther. 2021;11:639‐655.10.1007/s 40121-021-00519-2PMC 848175734591258 · doi ↗ · pubmed ↗
- 7Seib KL , Scarselli M , Comanducci M , Toneatto D , Masignani V . Neisseria meningitidis factor H‐binding protein f Hbp: a key virulence factor and vaccine antigen. Expert Rev Vaccines. 2015;14:841‐859.25704037 10.1586/14760584.2015.1016915 · doi ↗ · pubmed ↗
- 8Serruto D , Bottomley MJ , Ram S , Giuliani MM , Rappuoli R . The new multicomponent vaccine against meningococcal serogroup B, 4C Men B: immunological, functional and structural characterization of the antigens. Vaccine. 2012;30(Suppl. 2):B 87‐B 97.22607904 10.1016/j.vaccine.2012.01.033PMC 3360877 · doi ↗ · pubmed ↗