Inflammatory Myofibroblastic Tumor of the Lung: A Report of a Rare Case
Jayashree Bhawani, Samarth Shukla, Sourya Acharya

TL;DR
This paper reports a rare case of a lung tumor called inflammatory myofibroblastic tumor in a 53-year-old woman, highlighting its unusual presentation and diagnostic challenges.
Contribution
The novelty lies in presenting a rare case of PIMT in an older adult, expanding understanding of its clinical variability.
Findings
PIMT typically affects children and young adults but can occur in older individuals.
Diagnosis relies on histopathology, immunohistochemistry, and molecular testing.
Surgical resection is the primary treatment with a generally favorable prognosis.
Abstract
The uncommon and mysterious pulmonary inflammatory myofibroblastic tumor (PIMT) primarily affects children and young people. PIMT is characterized by the proliferation of myofibroblastic spindle cells mixed with inflammatory cells. It can resemble both benign and malignant disorders, both radiographically and clinically. PIMT typically manifests as a solitary lung tumor. The genesis of the tumor is linked to genetic anomalies, including those related to the ALK gene (anaplastic lymphoma kinase); nonetheless, some cases are not ALK-positive, indicating genetic variability. Clinically, patients may have non-specific symptoms such as cough, chest pain, or hemoptysis, or they may not exhibit any symptoms at all. In these cases, imaging tests may unintentionally reveal unrelated conditions. From a histopathological perspective, PIMT is characterized by a heterogeneous cellular makeup,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
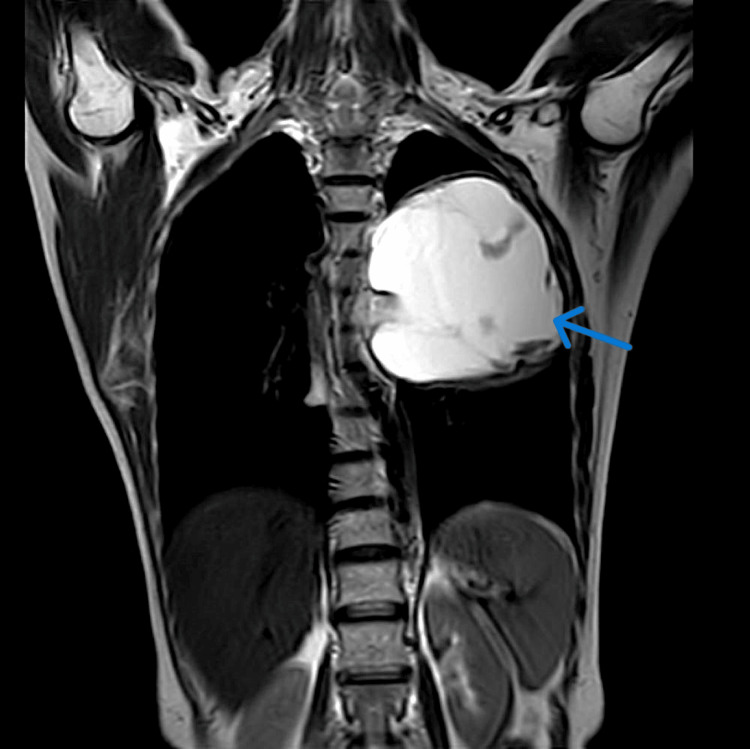 Figure 1
Figure 1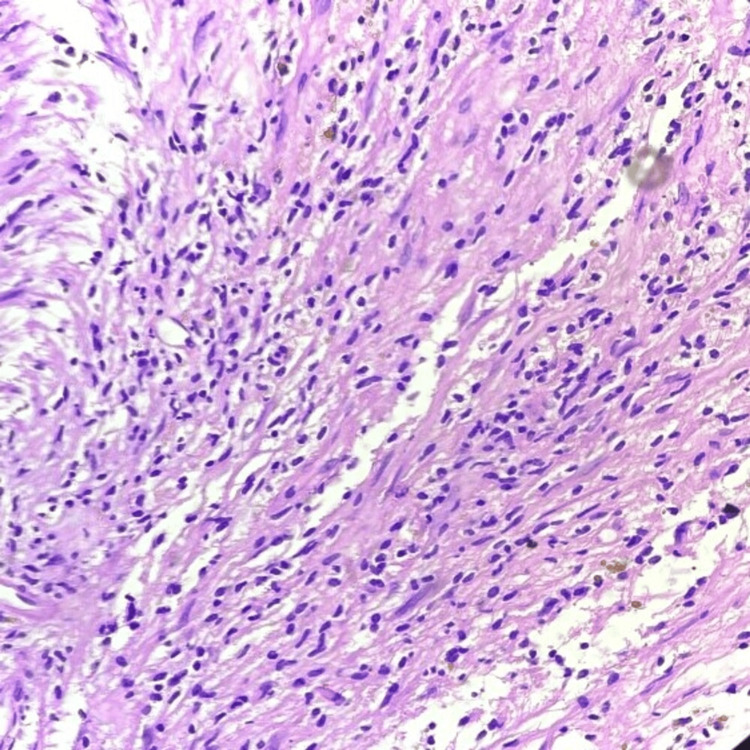 Figure 2
Figure 2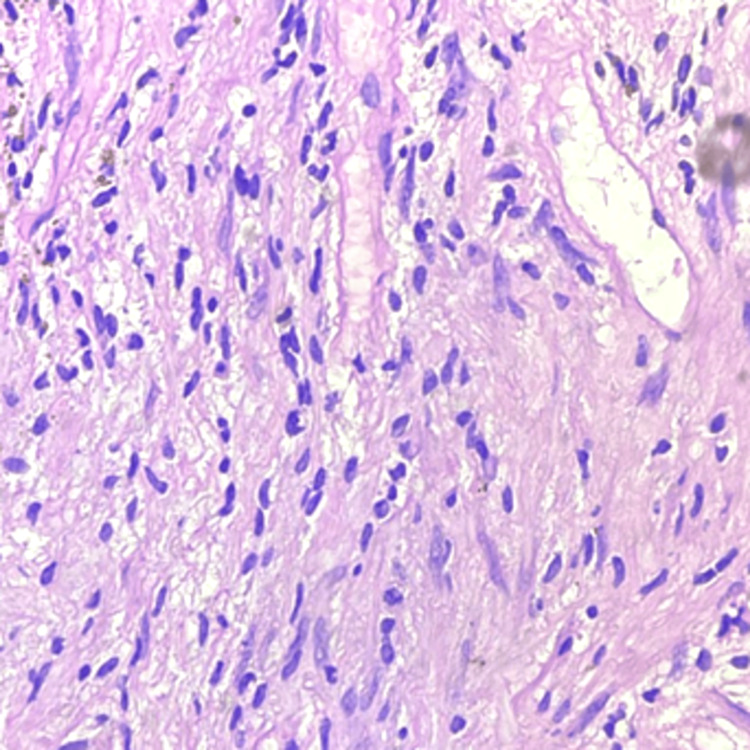 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsIgG4-Related and Inflammatory Diseases · Neuroendocrine Tumor Research Advances · Soft tissue tumor case studies
Introduction
The term "inflammatory pseudotumor" (IPT) describes a group of neoplastic and non-neoplastic entities with similar histological features, such as a prominent and usually persistent inflammatory infiltrate and spindle cell growth without cytology.
In the general category of inflammatory pseudotumors, the inflammatory myofibroblastic tumor (IMT) emerged in 1990, exhibiting unique clinical, pathological, and molecular characteristics [1]. Although Brunn initially reported it in 1939, its etiology is still unknown. IMTs are now categorized as uncommon intermediate-grade neoplasms with minimal metastatic potential and a high recurrence rate following excision [2,3].
IMTs account for 0.04% to 0.1% of all pulmonary neoplasms. They are more common in children and non-smoking adults [4,5]. Its clinical behavior is quite variable and is usually benign in nature. However, the possibility of metastasis cannot be denied in several cases. Diagnosis is often challenging and, many times, only possible after resection of the tumor. IMT is highly uncommon in adult females and rarely documented in the lungs. This study presents a highly uncommon case of a patient with pulmonary IMTs.
Case presentation
A 53-year-old married female presented to our healthcare center in February 2024 with complaints of breathlessness accompanied by chest discomfort and pain, which she had been experiencing for 10 days. There were no apparent symptoms of an irritating dry cough or sputum throughout this period. She was given symptomatic treatment and was sent back, but her condition did not improve. She came to the respiratory medicine outpatient department (OPD) after three days, and radiological investigations were performed.
A homogenous opacity across the entire upper lobe of the left lung was visible on the chest X-ray. An MRI was done to diagnose the lesion, revealing evidence of a well-defined hyperintense lesion, probably of neoplastic etiology (Figure 1). A biopsy was performed on the lesion at a private healthcare center. Biopsy reports were suggestive of adenocarcinoma of the lung. The patient reported to our cancer hospital for further treatment. After screening all her reports in a tumor board meeting, a decision to perform surgery was taken. A left pneumonectomy was performed as a last option for both therapeutic and diagnostic reasons. Grossly, the tumor was 7 x 3.2 x 2 cm in size, having a firm consistency. The tumor was grossly whitish and appeared homogeneous. Microscopy showed an increase in lymphocytes, plasma cells, eosinophils, and regular spindle cells arranged in fascicles (Figures 2, 3). A histopathological diagnosis of an IMT was given, and to confirm the diagnosis, immunohistochemistry was performed. ALK1 and SMA (smooth muscle actin) were detected positively by immunohistochemical investigation. Nevertheless, S100 had no effect on the tumor cells. These findings supported the diagnosis of an IMT. The patient was kept under medical observation, and antibiotics were administered for her recovery. The patient was administered IV Augmentin (1.5 g, TDS) and ceftriaxone (1 g, BD) for a duration of seven days. Postoperatively, the patient remained stable and was transferred to the ward. Follow-up was advised to the patient after 21 days.
MRI image showing evidence of a well-defined T2 hyperintense lesion (blue arrow) in the upper lobe of the left lung.
Photomicrograph (10x magnification) showing spindle cells arranged in fascicular arrangement with a surrounding population of lymphocytes and plasma cells on hematoxylin and eosin stain.
Photomicrograph (40x magnification) showing inflammatory cells with enlarged cells having spindle-shaped nuclei on hematoxylin and eosin stain.
Discussion
According to the latest WHO categorization, IMTs are now classified as tumors with intermediate biological potential. The tendency for local recurrence and the negligible chance of distant metastasis are cited as the causes. The anatomical site determines the recurrence rate, which ranges from 2% for lung-confined tumors to 25% for extrapulmonary lesions. The spleen, lymph nodes, esophagus, stomach, salivary glands, breast, epididymis, central nervous system, and soft tissues are among the additional locations that have been documented [6,7].
There is variability and non-specificity in the clinical and radiological presentations. Consequently, unless surgical excision is carried out, it is challenging to establish the diagnosis [8]. Radiological data are non-specific and subject to variation. In 87% of instances, a mass or nodular lesion with uniform boundaries is found. Its diameter is between 1 and 6 cm. Typically, nodules are solitary but can occasionally evolve into multiples [3].
Vimentin and SMA reactivity was demonstrated by immunohistochemistry. In slightly more than half of the instances with cytoplasmic staining, immunohistochemical positivity for ALK is visible; it is less common near the nuclear membrane [9].
Histopathology generally shows a mixed spectrum of spindled cells arranged in fascicles and having myofibroblastic differentiation. Tumor cells exhibit a storiform pattern [10]. Differentiating IMT's compact spindle cell pattern from its many histological siblings is a laborious task. A noticeable inflammatory infiltration is seen in sporadic spindle cell sarcomas, spindle cell melanomas, and sarcomatoid carcinomas, which may only exhibit modest cytological atypia. In contrast, plasma cells typically do not constitute a significant portion of the inflammatory infiltrate in other forms of tumors.
The most common pattern observed has at least focally prominent nuclear hyperchromasia, atypical mitoses, necrosis, or vascular invasion, which are highly uncommon in IMT [11,12]. The histology traits listed above were consistently present in this case in addition to the IMT characteristics.
However, the outcome after complete resection is excellent; chemotherapy may be used for multifocal, invasive lesions. It can also be used in cases of local recurrence. Patients should be kept under observation and tracked at short intervals by bronchoscopy following the complete resection and thoracic tomography for any potential recurrence [3].
Conclusions
Ultimately, the example of the lung's IMT highlights the intricacy and difficulties in diagnosing this uncommon disease. IMTs are benign but may be aggressive tumors, and a proper diagnosis frequently necessitates a high index of suspicion. A final diagnosis for our patient required a review of histological slides, radiological results, and clinical presentation. Surgery was the mainstay of the treatment plan, and it was successful in completely removing the tumor and reducing symptoms.
The significance of taking IMT into account when making a differential diagnosis for pulmonary masses is demonstrated by this instance, especially in patients who have non-specific respiratory symptoms. The importance of histology cannot be overstated, and the diagnosis can be further reinforced by immunohistochemistry staining, which identifies distinctive markers. Patients with pulmonary IMTs often have a good prognosis after complete surgical resection; nevertheless, ongoing surveillance is required to watch for metastasis or recurrence. The case study adds to the increasing corpus of knowledge on IMTs by highlighting the importance of interdisciplinary approaches and awareness in ensuring prompt diagnosis and treatment. Improving patient outcomes and deepening our knowledge of this uncommon ailment will benefit from additional studies into the pathophysiology and best management practices for IMTs.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Inflammatory myofibroblastic tumours: where are we now?J Clin Pathol Gleason BC Hornick JL 4284376120081793815910.1136/jcp.2007.049387 · doi ↗ · pubmed ↗
- 2What do we know about inflammatory myofibroblastic tumors? - a systematic review Adv Med Sci Siemion K Reszec-Gielazyn J Kisluk J Roszkowiak L Zak J Korzynska A 129138672022 https://doi.org/10.1016/j.advms.2022.02.0023521920110.1016/j.advms.2022.02.002 · doi ↗ · pubmed ↗
- 3Inflammatory myofibroblastic tumor: a rare tumor of the lung Eur Clin Respir J İçmeliÖS Alpay LA GündoğuşB Türker H Şen A 12014 https://doi.org/10.3402/ecrj.v 1.2539010.3402/ecrj.v 1.25390 PMC 462971826557237 · doi ↗ · pubmed ↗
- 4Inflammatory myofibroblastic tumor of the lung Adv Respir Med Khatri A Agrawal A Sikachi RR Mehta D Sahni S Meena N 2735862018 https://doi.org/10.5603/ARM.2018.00072949041910.5603/ARM.2018.0007 · doi ↗ · pubmed ↗
- 5Inflammatory myofibroblastic tumor of lung (pseudotumor of the lung)Indian J Radiol Imaging Mahale A Venugopal A Acharya V Kishore MS Shanmuganathan A Dhungel K 207210162006
- 6Inflammatory pseudotumors of the lung in a child (Article in French)J Pediatr Puericulture Almadi A Rami M Khattala K 6971242011
- 7Inflammatory myofibroblastic tumor of the lung: a rare entity Respir Med Case Reports Braham Y Migaou A Njima M 25312020 https://doi.org/10.1016%2Fj.rmcr.2020.10128710.1016/j.rmcr.2020.101287 PMC 768326233251105 · doi ↗ · pubmed ↗
- 8Diagnostic and therapeutic challenges of a large pleural inflammatory myofibroblastic tumor Case Rep Pulmonol Loeffler-Ragg J Bodner J Freund M Steurer M Uprimny C Zelger B Kähler CM 10219620122012 https://doi.org/10.1155/2012/1021962334644310.1155/2012/102196 PMC 3549377 · doi ↗ · pubmed ↗