A Case Report of Pulmonary Alveolar Proteinosis Masquerading as Respiratory Distress Syndrome in Preterm Neonates
Aditi Rawat, Sagar Karotkar, Mahaveer S Lakra, Snehlata Hingway, Revatdhamma Meshram, Amar Taksande

TL;DR
A preterm baby showed signs of respiratory distress syndrome but was later found to have a rare condition called pulmonary alveolar proteinosis, which is hard to diagnose and treat.
Contribution
This case highlights the diagnostic challenge of PAP in neonates and its resemblance to RDS.
Findings
The neonate's condition was misdiagnosed as RDS but was later confirmed as PAP via postmortem lung biopsy.
PAP is caused by abnormal surfactant protein and lipid accumulation in the alveoli, impairing gas exchange.
Treatment options for neonatal PAP are limited, with whole-lung lavage showing limited success.
Abstract
A rare and challenging case of a preterm neonate with clinical and radiological signs of respiratory distress syndrome (RDS) since the first hour of life but was refractory to its standard treatment regimes like surfactant therapy and ventilation. Postmortem lung biopsy led us to the diagnosis of congenital pulmonary alveolar proteinosis (PAP). It occurs due to the aggregation of abnormal surfactant proteins and lipids in the alveoli, which hampers gas diffusion across the alveoli. It presents as respiratory distress at birth, and its diagnosis is often missed due to its resemblance with RDS. Although the exact etiology remains elusive, mutations in genes encoding surfactant and granulocyte-macrophage colony-stimulating factor (GM-CSF) pathway components have been implicated in the pathogenesis of PAP. Treatment options are limited and only supportive. Among all these, whole-lung lavage…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
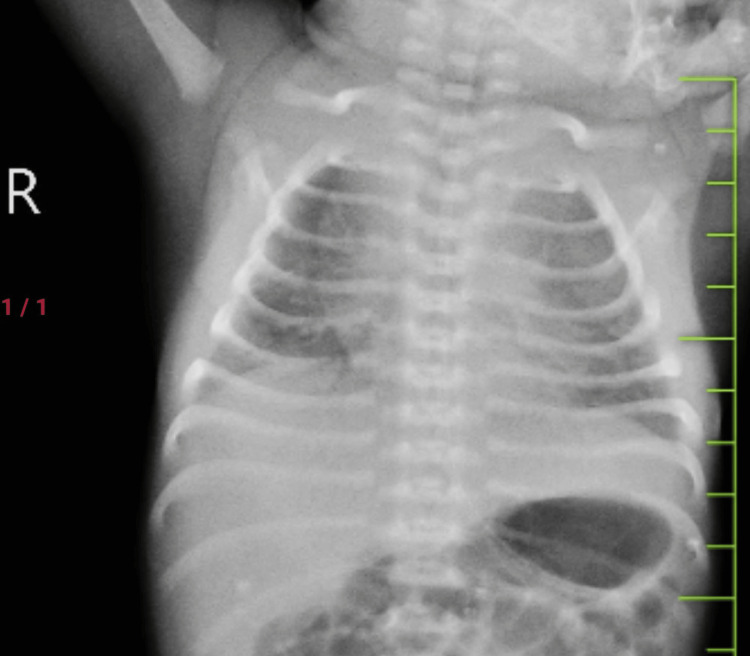 Figure 1
Figure 1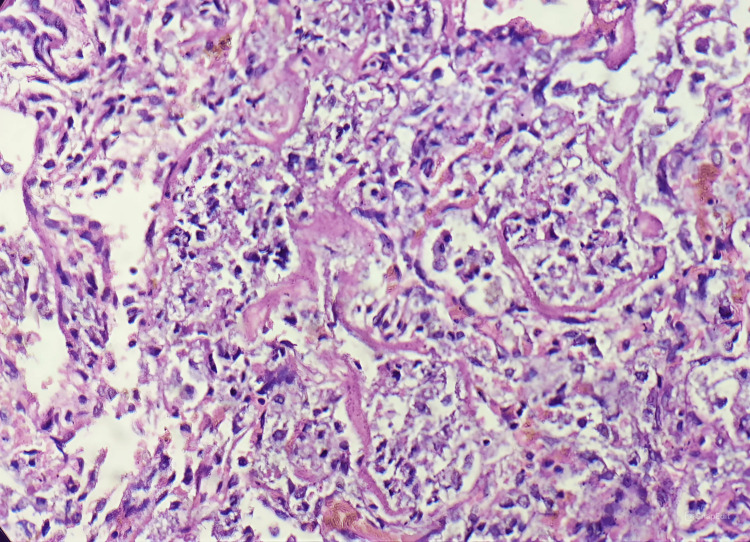 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeonatal Respiratory Health Research · Congenital Diaphragmatic Hernia Studies · Neuroscience of respiration and sleep
Introduction
Neonatal pulmonary alveolar proteinosis (PAP) is a rare and potentially life-threatening condition that affects newborns. It is distinguished by the collection of a periodic acid-Schiff (PAS)-positive diastase-resistant amorphous proteinaceous material in the alveoli leading to respiratory insufficiency [1]. Two types of PAP have been described; congenital PAP which has a fulminant course and is usually fatal, and other one is late-onset PAP which is less severe and mimics the adult form [2]. The clinical presentation at birth is nonspecific ranging from asymptomatic to progressive respiratory failure. The diagnosis is confirmed by lung biopsy and bronchoalveolar lavage [3]. Treatment options are limited with poor success rates including whole-lung lavage, lung transplantation, administration of granulocyte-macrophage colony-stimulating factor (GM-CSF), and gene therapy [4]. Here, we present a case of a severe form of congenital PAP presenting at birth as respiratory distress refractory to standard treatment.
Case presentation
A 1.3 kg female child was born to a 28-year-old multigravida mother with a bad obstetric history at 28 weeks of gestation by cesarean section in view of fetal distress. Antenatal history revealed four previous neonatal deaths out of which one was stillbirth and the rest all succumbed within 24 hours of life. All previous pregnancies were preterm births, and the babies had respiratory distress at birth. The mother had a unicornuate uterus which was hypothesized to be the cause for premature deliveries. There was a history of third-degree consanguinity present. The antenatal period was uneventful till 28 weeks when the patient started experiencing preterm labor for which she was admitted, antenatal steroid coverage with intravenous (IV) dexamethasone was given, and owing to fetal compromise, she was taken up for emergency cesarean.
The baby had severe respiratory distress at birth requiring delivery room continuous positive airway pressure (CPAP). In the neonatal intensive care unit (NICU), the baby was kept on CPAP support with a fraction of inspired oxygen (FiO2) requirement of 30% and a positive end-expiratory pressure (PEEP) of 5. Total parenteral nutrition and first-line antibiotics were initiated. The baby had a Silverman Anderson score of 5 with a chest x-ray suggestive of low lung volume and bilateral homogenous ground glass appearance suggestive of respiratory distress syndrome (RDS) as seen in Figure 1.
Low lung volume with bilateral homogenous ground glass opacities in lung fields suggestive of respiratory distress syndrome
In view of the increasing Fio2 requirement of >30%, Neosurf was administered within two hours of life as standard treatment protocol for RDS. In view of persistent respiratory distress, the baby was intubated by the fourth hour of life on a conventional ventilator. A second dose of surfactant, i.e., Neosurf, was administered. After 24 hours, the respiratory distress settled, and the baby was weaned off to CPAP by 36 hours of life.
After 12 hours of extubation, the respiratory distress increased mandating re-intubation. Septic screen came out negative. There was persistent desaturation with blood gas suggestive of respiratory acidosis for which the conventional ventilation setting was increased gradually to a maximum of Fio2 of 100%, PEEP of 6, and peak inspiratory pressure (PIP) of 24. The baby was shifted to high-frequency ventilation thereafter. The baby failed to show improvement despite adequate ventilation and succumbed due to refractory respiratory failure.
Postmortem lung biopsy was done suspecting surfactant deficiency due to previous neonatal deaths and nonresponse to conventional treatment with maximum ventilatory support. The biopsy was suggestive of diffuse alveolar damage with PAS-positive hyaline proteinaceous material lining the alveoli and preserved lung architecture suggestive of congenital alveolar proteinosis as depicted in Figure 2.
Lung biopsy showing the alveoli filled with hyaline matter and preserved lung architecture characteristic of pulmonary alveolar proteinosis
Due to financial constraints, the genetic mutation workup could not be carried out for this case. The parents were advised for genetic counselling before planning next pregnancy.
Discussion
Congenital alveolar proteinosis is a rare genetic disorder with a reported prevalence of 0.1 per 100,000 with a characteristic deposition of abnormal surfactant protein within the alveoli [4]. Surfactant is synthesized by type II alveolar cells which act by reducing surface tension in the alveoli, thus preventing its collapsing during exhalation.
The exact cause of congenital PAP remains unclear although the widely accepted etiology is a mutation in surfactant protein B or C leading to intra-alveolar deposition of these abnormal nonfunctioning proteins as seen in our case on lung biopsy. Recently, a mutation in alpha or beta chains of GM-CSF has been identified in congenital PAP, although it is usually seen in adult forms of PAP [5].
A family history of similar respiratory issues in sibling or consanguineous marriage points toward an autosomal recessive transmission [4]. A history of consanguinity is an important clue which is usually present in 35% of the cases and was present in our case too [6].
According to available literature, there is a significant time gap between the onset of symptoms and the diagnosis of a patient of PAP which is around 4.5 months [6].
The symptoms are usually nonspecific presenting as growth retardation and progressive dyspnea in the majority of cases [7]. In our case, the presentation was very aggressive as symptoms were seen immediately after birth.
The diagnostic modalities available are X-rays, high-resolution computed tomography (CT) thorax, and bronchoalveolar lavage (BAL). CT thorax will show thickened septa with a characteristic crazy pavement pattern [8]. BAL will show a milky appearance of the washed fluid and a PAS-positive substance in the alveolar macrophage [3]. In our case, as the diagnosis was made on postmortem analysis, similar finding was seen in lung biopsy histology.
Treatment options are very limited and aimed at improving respiratory function and providing supportive care. A whole-lung lavage is the only intervention that is used widely and associated with long-term improvement [4]. The goal is to remove the proteinaceous material in alveoli and restore the gas exchange at the alveolar-capillary barrier, although this is of limited use in newborns because the use of large endotracheal tubes is associated with technical problems [9]. Other available less-used modalities are lung transplantation, gene therapy, GM-CSF administration.
Without lung transplantation, the mortality of congenital PAP is almost 100% [10]. Late-onset disease has better prognosis. Disease-specific five-year survival rates are 88% and 80% of deaths occur within the first year [7].
Conclusions
PAP is a very rare disease causing respiratory distress; due to variations in the severity of the disease, and the pediatrician’s nonfamiliarity with PAP, its diagnosis is usually delayed or missed. It should be considered in cases with consanguinity, positive family history, and nonresponsiveness to standard treatment. Diagnosis can be confirmed on histopathologic analysis of samples from BAL or lung biopsy. Whole-lung lavage can be tried to improve survival and severe cases may need lung transplantation. The overall prognosis is poor and genetic counselling should be planned for subsequent pregnancies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Pulmonary alveolar proteinosis in a newborn Pediatr Pulmonol Schumacher RE Marrogi AJ Heidelberger KP 17818271989267794410.1002/ppul.1950070312 · doi ↗ · pubmed ↗
- 2Pulmonary alveolar proteinosis in children: a case series J Res Med Sci Tabatabaei SA Karimi A Tabatabaei SR Radpay B Jadali F Shiva F Jahromy MH 120124152010 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC 3082791/21526069 PMC 3082791 · pubmed ↗
- 3Pulmonary alveolar proteinosis in children Paediatr Respir Rev de Blic J 316322520041553125710.1016/j.prrv.2004.07.001 · doi ↗ · pubmed ↗
- 4Congenital pulmonary alveolar proteinosis Case Rep Pediatr Hammami S Harrathi K Lajmi K Hadded S Ben Meriem C Guédiche MN 764216201320132371040310.1155/2013/764216 PMC 3655498 · doi ↗ · pubmed ↗
- 5Long-term follow-up and treatment of congenital alveolar proteinosis BMC Pediatr Griese M Ripper J Sibbersen A 721120112184903310.1186/1471-2431-11-72PMC 3175167 · doi ↗ · pubmed ↗
- 6Pulmonary alveolar proteinosis: case report of rare diffuse lung disorder in pediatric age group Indian J Respir Care Gandhi V Kadam S 169172112022
- 7Pulmonary alveolar proteinosis Indian Pediatr Garg G Sachdev A Gupta D 521523462009 https://pubmed.ncbi.nlm.nih.gov/19556662/19556662 · pubmed ↗
- 8Pulmonary alveolar proteinosis: clinical aspects and current concepts on pathogenesis Thorax Shah PL Hansell D Lawson PR Reid KB Morgan C 67775520001060780510.1136/thorax.55.1.67PMC 1745595 · doi ↗ · pubmed ↗