Familial 46, XY Disorder of Sexual Development identified in a Ph+BCR::ABL1P210+ Acute Lymphoblastic Leukemia septuagenarian female with RCBTB2::LPAR6 fusion gene: a case report
Lingling Wang, Conglin Xi, Xinyu Zheng, Yongfen Huang, Hao Xu, Yuqing Miao, Yuexin Cheng

TL;DR
An elderly woman with a male karyotype and leukemia is found to have a rare disorder of sexual development, along with a rare fusion gene and a family history of similar issues.
Contribution
This is the first reported case of familial 46, XY DSD with Ph+ ALL and the RCBTB2::LPAR6 fusion gene in an elderly patient.
Findings
The patient had a male karyotype despite being identified as female, with a rare RCBTB2::LPAR6 fusion gene and Ph+ ALL.
Her sister also had a male karyotype and similar reproductive issues, indicating a possible familial DSD.
This case highlights the under-recognized risk of hematologic malignancies in individuals with 46, XY DSD.
Abstract
Familial 46, XY Disorder of Sexual Development (DSD) was discovered in a Ph+, BCR::ABL1P210+ Acute Lymphoblastic Leukemia (ALL) female with RCBTB2::LPAR6 fusion gene. Siblings developing 46, XY DSD are extremely rare. Patients with 46, XY DSD have much higher rates of gonadal cancers. Nevertheless, the incidence of hematologic malignancies in patients with DSDs has received little attention. RCBTB2::LPAR6 is a rarely reported fusion gene in ALL. Herein, we report a rare case of a newly diagnosed Ph+, BCR::ABL1P210+ ALL patient who was 77 years old and female by social sex. Whole Exome Sequencing (WES) and RNA sequencing revealed TET2 and NF1 mutations in addition to a rarely reported RCBTB2::LPAR6 fusion gene and 17 other genes with uncertain clinical significance. The patient was surprisingly found to have a male karyotype. On ultrasound, neither the uterus nor the ovaries were…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
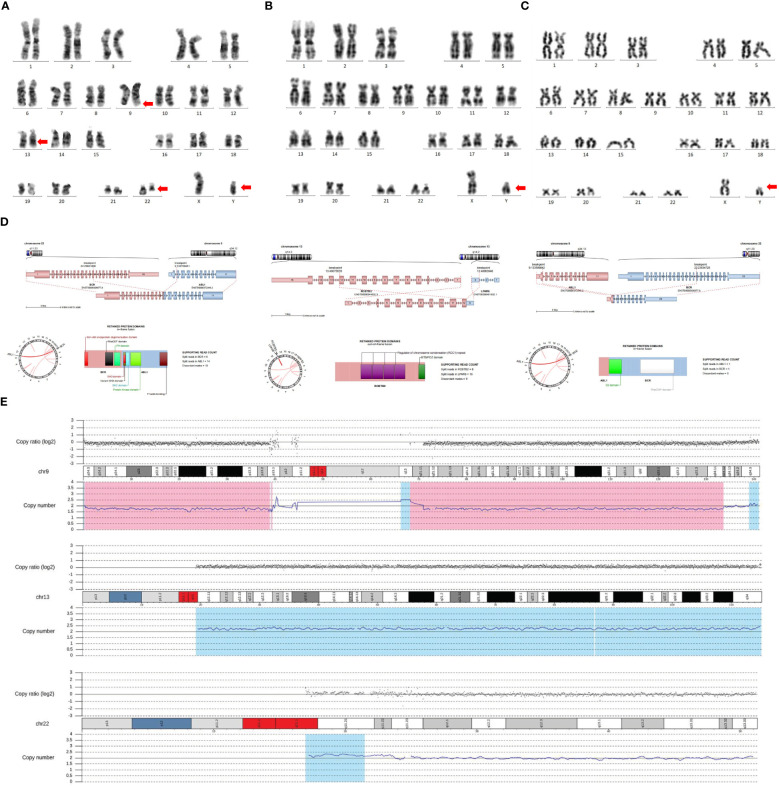 Figure 1
Figure 1 Figure 2
Figure 2| No | Mutant Gene | Chromosome Location | Nucleotide and Amino Acid Changes | Allelic Ratio |
|---|---|---|---|---|
| 1 | ARID1A | chr1:27087947 | c.2234G>A | 0.397 |
| 2 | CHL1 | chr3:391167 | c.974G>A | 0.417 |
| 3 | HACE1 | chr6:105231979 | c.1548G>A | 0.456 |
| 4 | ANKRD17 | chr4:73963838 | c.4973C>T | 0.374 |
| 5 | CYBA | chr16:88717415 | c.7C>T | 0.527 |
| 6 | DNAH12 | chr3: 57391391 | c.6565C>A | 0.466 |
| 7 | DNAH12 | chr3:57432425 | c.3787–9T>C | 0.541 |
| 8 | FANCG | chr9:35075597 | c.1298G>A | 0.518 |
| 9 | FAT2 | chr5:150948447 | c.46G>T | 0.595 |
| 10 | HKDC1 | chr10:71026409 | c.2650C>G | 0.452 |
| 11 | L4R | chr16:27353547 | c.176G>A | 0.507 |
| 12 | NFATC4 | chr14:24845662 | c.2408G>C | 0.519 |
| 13 | PDE4DIP | chr1:144868044 | c.5881A>G | 0.232 |
| 14 | SDK2 | chr17:71503692 | c.109G>T | 0.425 |
| 15 | SDK2 | chr17:71503707 | c.94C>G | 0.438 |
| 16 | TENM3 | chr4:183658049 | c.3056A>G | 0.509 |
| 17 | TSC2 | chr16:2108740 | c.849–8A>G | 0.552 |
| Reference | Age at diagnose | Symptoms and physical examinations | Mutation etiology |
|---|---|---|---|
| Berg et al, 1987 ( | 20/18 years old | Primary amenorrhea, retarded breast development, scanty axillary and pubic hair growth. | gonadal dysgenesis |
| Shahid et al, 2007 ( | 23/27 years old | Primary amenorrhea, and clitoromegaly without labial fusion. | SRY mutations |
| Bisceglia et al, 2008 ( | 4 years old/ | Female external genitalia with a shortened vagina, normal for age breast development, absent pubic and axillary hair, no uterus, no fallopian tubes, and no ovaries. | complete androgen insensitivity |
| Nichols et al, 2008 ( | 17/17 years old | Primary amenorrhea, without appropriate breast and pubic hair development. | complete androgen insensitivity |
| Sharma et al, 2010 ( | 18 | Axillary and pubic hair was scanty, with no hair growth, normal female genitalia, labia major, labia minor, and clitoris, with prominent bilateral inguinal swelling. | androgen insensitivity |
| Omrani et al, 2011 ( | 28/19/18 years old | Hirsutism and deepening of the voice, increased musculature, enlarged clitoris (6–7 cm), lack of breast development, and primary amenorrhea. | HSD17B3 mutations |
| Fabbri et al, 2014 ( | S1: 9 months | S1: ambiguous genitalia | NR5A1 mutations |
| Paris et al, 2017 ( | 26/23 years old | Primary amenorrhea, without breast development and scanty pubic hair. | homozygous DHH mutations |
| Banoth et al, 2017 ( | 29/23/17 years old | primary amenorrhea | gonadal dysgenesis |
| Ilaslan et al, 2018 ( | S1: 17 years old | S1: lack signs of puberty and menstruation, female-type external genitalia, oviducts, a normal prepubertal vagina, and uterus | gonadal dysgenesis, STARD8 |
| Putri et al, 2022 ( | 19/17/15 years old | Late menarche, elementary breast development, and little axillary hair. The external genitals consisted of the vulva and major and minor labia. Clitoromegaly was present with a short ( | type 2 5-α reductase deficiency |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRenal and related cancers · Acute Lymphoblastic Leukemia research · Cancer-related gene regulation
Background
Ph+ ALL is rare in children, with an incidence of 2%-5%. However, it is the most common genetic subgroup in adult ALL (1), with an overall incidence of 20%-25%, and the incidence increases with age, with patients over 50 years of age accounting for more than 50%. Ph+*BCR::ABL1^P210+^
- ALL is a rare type of ALL in the elderly. RCBTB2::LPAR6 is a rarely reported fusion gene in Ph+*BCR::ABL1^P210+^
- ALL.
DSD refers to a congenital chromosomal, gonadal, and phenotypic sex-linked developmental anomaly or mismatch that includes a variety of inborn metabolic anomalies and deformities, mostly marked by abnormalities of the external genitalia. There are three types of sex developmental anomalies: 46, XX DSD, 46, XY DSD, and sex chromosome DSD (2). Among these, 46, XY DSD has a karyotype of 46, XY, and may have a Müllerian duct structure. The external genitalia may be either normal male genitalia, ambiguous external genitalia, or even completely feminized (3). Familial 46, XY DSD is extremely rare, and studies of siblings with concurrent 46, XY DSD are uncommon, with only a few case reports to date. There are few studies on the prevalence of neoplasms, particularly hematologic malignancies, among DSD patients, especially the familial 46, XY DSD.
Herein, we report a rare case of a newly diagnosed Ph+*BCR::ABL1^P210+^
- ALL patient who was 77 years female by social sex but had a male karyotype. A rarely reported RCBTB2::LPAR6 fusion gene was discovered. After gathering a complete family history and marriage history, it was unexpectedly found that the patient’s sister who was 73 years was also a patient of 46, XY DSD.
Although both patients were in their seventies and no longer in need of childbearing, they were still puzzled about the cause of their infertility. With the consent of the patient and her family members, and the approval of the Medical Ethics Committee of The First People’s Hospital of Yancheng (Ethics Approval No. 2023-k-195), we initiated the study. Elderly patients with DSD do not receive much attention and support from society, partly due to the backwardness of medical testing technology in the past decades, and partly due to the poor accessibility of medicine for the general population. With the maturity of WES and RNA sequencing, more potential DSD patients may be diagnosed, and this population needs the attention of clinicians, not only limited to medical treatment but also more humanistic care and social support. This was our original intention in reporting this case.
Case presentation
In April 2023, a 77-year-old female presented to our hospital complaining of fatigue, bone pain, and skin petechiae. The patient had a history of coronary atherosclerotic heart disease, type 2 diabetes, and hypertension. A physical examination revealed no lymphadenopathy, hepatomegaly, or splenomegaly. She was found to have a WBC of 100 × 10^9^/L with 9% blasts, Hb of 9.1 g/dL, and platelet count of 14 × 10^9^/L. Serum aspartate aminotransferase was elevated to 145.0 U/L, albumin was 36.4 g/L, lactate dehydrogenase was elevated to 5999.0 U/L, and creatinine was 47.3 μmol/L. Serum electrolytes were normal. N-terminal brain natriuretic peptide was elevated to 881 pg/ml. The bone marrow biopsy revealed 28.5% blasts (CD13+, CD19+, CD20+, CD10+, CyCD79a+, CD34+, HLA-DR+). Fluorescence in situ hybridization (FISH) and reverse transcriptase polymerase chain reaction (RT-PCR) were positive for the presence of BCR::ABL1 (coding for a 210-kDa protein). G-banding analysis of bone marrow cells indicated the karyotype 46, XY, t (9;22)(q34;q11.2)[10]/47, idem, +der(22)t (9;22)[1]/46, idem, del(13)(q12q14)[1] (Figure 1A).
Testing techniques include G-banding technique, WES, and RNA sequencing. (A) G-banding analysis of bone marrow cells indicated the karyotype of 46, XY, t(9;22)(q34;q11.2)[10]/47, idem, +der(22)t(9;22)[1]/46, idem, del(13)(q12q14)[1]. (B) The chromosome analysis was performed with the patient’s peripheral blood, which still revealed a male karyotype. (C) The chromosomes of this patient’s sister were suggestive of a male karyotype. (D) WES and RNA sequencing indicated three fusion genes, including BCR::ABL1, ABL1::BCR, and RCBTB2::LPAR6. (E) WES and RNA sequencing indicated 46, XY DSD, with three additional chromosomal variants, del(9) (p24.3q34.12), dup(13) (q11q34), and dup(22) (q11.1q11.23). The genomic coordinates were chr9:g.190065_133643956del, chr13:g.19194200_115109878dup, and chr22:g.16962100_23627091dup, respectively.
The patient’s social sex was female, yet the chromosomes were of the male karyotype. We re-examined the patient’s peripheral blood for chromosomal verification, which was still suggestive of a male karyotype (Figure 1B). On ultrasound, neither the uterus nor the ovaries were discernible. A detailed family and marital history revealed that the patient had undergone surgery at an early age for an unexplained inguinal mass. She had slow pubertal development, scanty menstruation, and few overtly feminine characteristics. She had three previous marriages, but none succeeded in getting pregnant. The patient did not seek an early diagnosis and treatment because of medical inaccessibility when she was young and a lack of knowledge about medicine. Examination of the external genitalia revealed an enlarged clitoris. Laboratory analysis revealed only 5.47 (range: 1030) pg/ml of estradiol, a significantly reduced quantity. Blood testing revealed that male testosterone levels were normal, at 0.2 (range: 0.10.75) ng/mL. In light of this, we speculate that this patient might be an unusual case of Ph+*BCR::ABL1^P210+^
- ALL combined with 46, XY DSD. WES and RNA sequencing were performed to determine whether the patient had DSD-related mutations. NF1 [11.2%; NM_001042492: c.7194C>A(p.Tyr2398Ter)] and TET2 [39.9%; NM_001127208: c.3743T>C(p.Leu1248Pro)], as well as 17 other mutations, were detected (Table 1). In addition to the BCR::ABL1 fusion gene, the ABL1::BCR fusion gene, and a novel RCBTB2::LPAR6 fusion gene were also discovered (Figure 1D). Copy Number Variation Sequencing (CNV-Seq) confirmed that the patient’s sex chromosomes were XY, which was inconsistent with social gender, along with three autosomal variants (del(9) [p24.3q34.12), dup(13) (q11q34), and dup(22) (q11.1q11.23)] (Figure 1E). Her sister, a 73-year-old female by social sex, who had amenorrhea in adolescence and was unable to conceive, had the same experience. When we karyotyped the patient’s sister, we discovered, to our surprise, that she too had a male karyotype (Figure 1C). However, she did not suffer any gonadal or hematologic malignancies.
Table 1: Whole Exome Sequencing and RNA sequencing a identified a total of 17 variants with undetermined clinical significance.
We initially prescribed dexamethasone 10 mg/d to reduce the tumor load. Fortunately, the patient did not experience tumor lysis syndrome. The patient initially displayed doubt about the diagnosis, mistrust of the doctor, and refusal of family care. The patient had very low compliance and was agitated. After extensive deliberation and reassurance, the patient eventually consented to Imatinib 400 mg/d treatment. During the therapy, the patient developed pancytopenia combined with a severe lung infection, and ultimately died of the infection one month later. Figure 2 shows the treatment timeline of this patient.
The patient’s treatment timeline.
Discussion
DSD is a congenital disorder that presents as incongruence in an individual’s sex chromosomes, gonads, and/or anatomical sex (4). Cases of 46, XY DSD are generally sporadic, and familial aggregation is extremely rare. Typically, 46, XY DSD patients will have varying degrees of masculinization of the external genitalia. Structures from Wolffian and Müllerian ducts also develop to varying degrees. The presence of testes can be detected by imaging in the majority of patients. Whereas 46, XY DSD can be identified through prenatal diagnosis, a portion is diagnosed at birth because of atypical external genitalia. In patients with more pronounced feminization, the probability of recognition at an early stage is lower, and most of them are diagnosed with 46, XY DSD by the presence of primary amenorrhea as well as unexplained inguinal hernia presenting in childhood, delayed breast development during puberty, or by the inability to be fertile. There are two main causes of 46, XY DSD, a decrease in the secretion of androgens (e.g., testosterone or dihydrotestosterone) during fetal sex differentiation, and impaired action of androgens on target tissues throughout the life course. The identification of novel genotypes has been made possible by WES. WT1, GATA4, DHH, SOX9, NR5A1, MAP3K1, DHX37, and SRY are the key potential genes that have been identified in 46, XY DSD. DHCR7, STAR, CYP17, POR, B5, HSD3B2, HSD17B3, and CYP11A are candidate genes for steroidogenic enzyme defects. Siblings sharing 46, XY DSD are very rare. We identified 11 pairs of siblings after synthesizing the published case reports (Table 2) (5–15). These patients were usually identified when they were young or in their teens, typically through observing vulvar anomalies or amenorrhea in the proband, followed by identifying comparable disorders in the siblings, leading to a certain diagnosis. Due to the lack of long-term follow-up, the development of hematologic disorders, especially hematologic malignancies, was not observed among these sibling pairs.
According to recent research (16–18), these patients are more likely than the general population to develop gonadal malignancies, such as germ cell tumors, embryonal carcinomas, yolk sac tumors, choriocarcinomas, and teratomas (19, 20). In contrast, the incidence of other cancers is not increased. The incidence of cardiovascular diseases like hypertension, atherosclerotic vascular disease, or coronary artery disease is not noticeably higher in patients with 46, XY DSD. Agnethe Berglund et al (21) retrospectively analyzed a total of 123 with 46, XY DSD in Denmark from 1960 to 2012. They had a higher incidence of congenital malformations and endocrine and gastrointestinal disorders. Five patients developed anemia. By the end of the follow-up, no hematologic malignancies were observed. Beyond that, there are no more large-sample data on survival and comorbidities, especially hematologic disorders in 46, XY DSD patients.
Because of the intricate pathophysiology of 46, XY DSD, and the vast range of clinical manifestations, therapeutic therapy has mostly focused on and battled with regulating the disorder’s classification. The incidence of hematologic malignancies in DSD patients is unknown. To ascertain whether there are differences in incidence, mutation type, and prognosis compared with the general population, extensive clinical data analysis is still needed. From an epidemiologic point of view, more data are needed to support whether initiating epidemiologic screening of patients with DSD has positive sociologic and medical significance. There are still many aspects of previous clinical reports of DSD patients that need to be improved. First, the number of cases is small, and the reports are mainly from single centers, which lack the integration of data from multiple centers. Secondly, there is a lack of long-term clinical follow-up, and there are very few studies on elderly DSD patients. Whether there is a difference in the incidence of tumors in this group of patients compared with the general population needs to be further confirmed.
Patients with 46, XY DSD are more likely to experience depression, obsessive-compulsive disorder, and an antisocial disposition (22). Self-harm and suicidal thoughts may emerge in a portion of patients (23, 24). The patient was admitted to the hospital with dangerously low platelets and extremely high white blood cell counts. Potential adverse consequences included hyperviscosity and resultant brain and gastrointestinal hemorrhage. The patient initially displayed doubt about the diagnosis, mistrust of the doctor, and refusal of family care when she was told that she was a 46, XY DSD patient and was diagnosed with Ph+ALL. The patient had very low compliance, was agitated, and rejected intravenous chemotherapy. After extensive deliberation and reassurance, the patient finally agreed to the regimen of intravenous dexamethasone pretreatment for lowering tumor load and imatinib maintenance therapy. The patient, who had coronary artery disease, type 2 diabetes, and hypertension, passed away during therapy as a result of an uncontrollable infection. The patient’s sister, on the other hand, had for some time denied our medical follow-up because she found it difficult to accept that she was a DSD patient and that her older sister’s condition was deteriorating so quickly. She finally accepted the diagnosis after extensive family and medical counseling, and she decided to stick with her decision to identify as female. The patient’s sister is currently compliant, and we will be following her closely. The focus of the follow-up will be the presence of cardiovascular events, endocrine disorders, and the occurrence of tumors.
Gender selection, identification, and the ideal decision-making window are the challenges in the management of 46, XY DSD. The major purpose of sex determination is to align sex determination with the chosen sex to avoid increased risk of sexual anxiety. After receiving a conclusive diagnosis, the patient should be engaged in a thorough and objective discussion about gender determination, including the likelihood of normal sexual function, fertility, risk of gonadal malignancy, and the various potential options. Advice should then be given in the patient’s specific situation. High-quality, long-term psychosocial evidence is scarce on gender identity in 46, XY DSD patients. The optimum path of treatment for addressing problems with gender identity needs active collaboration between the patient, their family, and the doctor.
Conclusion
Studies of neoplasms in 46, XY DSD are limited to gonadal tumors, but the incidence and type of hematologic malignancies are incredibly uncommon. Siblings developing 46, XY DSD is extremely rare. We report the oldest patient diagnosed with 46, XY DSD. There have not been any reports of familial 46, XY DSD with a concurrent diagnosis of Ph+*BCR::ABL1^P210+^ *ALL and a rarely reported RCBTB2::LPAR6 fusion gene.
Nonetheless, given the scarcity of data on hematologic malignancies in DSD patients, widespread screening for hematologic abnormalities in this population is unlikely. What we can do is establish a database of patients with DSD with long-term follow-up to better monitor possible long-term comorbidities. Balancing the patient’s privacy and the social impact that long-term follow-up may have on the patient’s private life requires the cooperation of the patient’s family, the public, and the government.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by The First People’s Hospital of Yancheng. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
LW: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. CX: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. XZ: Conceptualization, Data curation, Investigation, Methodology, Software, Writing – original draft. YH: Conceptualization, Formal analysis, Investigation, Project administration, Software, Validation, Writing – review & editing. HX: Formal analysis, Funding acquisition, Project administration, Resources, Validation, Visualization, Writing – review & editing. YM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. YC: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1FoàR Chiaretti S. Philadelphia chromosome-positive acute lymphoblastic leukemia. N Engl J Med. (2022) 386:2399–411. doi: 10.1056/NEJ Mra 2113347 35731654 · doi ↗ · pubmed ↗
- 2Reyes AP León NY Frost ER Harley VR. Genetic control of typical and atypical sex development. Nat Rev Urol. (2023) 20:434–51. doi: 10.1038/s 41585-023-00754-x 37020056 · doi ↗ · pubmed ↗
- 3Hattori A Fukami M. Nuclear receptor gene variants underlying disorders/differences of sex development through abnormal testicular development. Biomolecules. (2023) 13:691. doi: 10.3390/biom 13040691 37189438 PMC 10135730 · doi ↗ · pubmed ↗
- 4Gomes NL Batista RL Nishi MY Lerário AM Silva T Ede Moraes Narcizo A. Contribution of clinical and genetic approaches for diagnosing 209 index cases with 46,XY differences of sex development. J Clin Endocrinol Metab. (2022) 107:e 1797–1797 e 1806. doi: 10.1210/clinem/dgac 064 35134971 · doi ↗ · pubmed ↗
- 5Berg FDKürzl R Hinrichsen MJ Zander J. Familial 46,XY pure gonadal dysgenesis and gonadoblastoma/dysgerminoma: case report. Gynecol Oncol. (1989) 32:261–7. doi: 10.1016/S 0090-8258(89)80046-0 2910791 · doi ↗ · pubmed ↗
- 6Shahid M Dhillon VS Hussain Z Masa JF Aslam M Raish M. Analysis of the SRY gene in two sex-reversed XY sisters identifies two new novel point mutations in the high mobility group box domain. Fertil Steril. (2008) 90:1199. doi: 10.1016/j.fertnstert.2007.11.062 18304538 · doi ↗ · pubmed ↗
- 7Bisceglia M Magro G Ben Dor D. Familial complete androgen insensitivity syndrome (Morris syndrome or testicular feminization syndrome) in 2 sisters. Adv Anat Pathol. (2008) 15:113–7. doi: 10.1097/PAP.0b 013e 318166 aa 3b 18418092 · doi ↗ · pubmed ↗
- 8Nichols JL Bieber EJ Gell JS. Case of sisters with complete androgen insensitivity syndrome and discordant Müllerian remnants. Fertil Steril. (2009) 91:932.e 15–8. doi: 10.1016/j.fertnstert.2008.09.027 18930210 · doi ↗ · pubmed ↗