The role of ribosomal DNA methylation in embryonic development, aging and diseases
Fei Yang, Xutong Guo, Yiming Bao, Rujiao Li

TL;DR
This review explores how ribosomal DNA methylation influences embryonic development, aging, and diseases across multiple species.
Contribution
The paper provides a comprehensive overview of rDNA methylation's role in biological processes and highlights current research gaps.
Findings
rDNA methylation is linked to epigenetic changes during embryonic development.
Methylation of rDNA is associated with aging and various diseases.
The review identifies gaps in understanding rDNA methylation's functional roles.
Abstract
The ribosomal DNA (rDNA) constitutes a remarkably conserved DNA sequence within species, located in the area of the nucleolus, and responsible for coding three major types of rRNAs (18S, 5.8S and 28S). While historical investigations into rDNA focused on its structure and coding capabilities, recent research has turned to explore its functional roles in various biological processes. In this review, we summarize the main findings of rDNA methylation with embryonic development, aging and diseases in multiple species, including epigenetic alterations, related biological processes and potential applications of rDNA methylation. We present an overview of current related research and identify gaps in this field.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
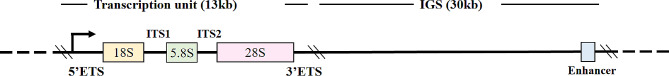 Figure 1
Figure 1- —http://dx.doi.org/10.13039/501100001809National Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLibraries, Manuscripts, and Books · Medieval and Early Modern Iberia · Historical Art and Architecture Studies
Introduction
Ribosomal DNA sequences are essential components of all eukaryotic genome sequences. In humans, the main rDNA unit spans approximately 43 kb, comprising a 13 kb transcribed region containing sequences encoding the 18S, 5.8S and 28S rRNAs, along with a 30 kb intergenic spacer (IGS) [1] (Fig. 1). Additionally, rDNA comprises a separate cluster of approximately 2.2 kb repeated genes that form the 5S rRNA [2, 3]. The critical components of IGS encompass upstream control elements (UCE) and core promoter elements (CPE), as well as other regulatory regions. rDNA is the only DNA present within the nucleolar volume [4], arranged in tandem repeat clusters in the nucleolar organizer regions (NORs), on the short arms of acrocentric chromosomes. rDNA gene sequences are highly conserved within species, characterized by high repeatability, high transcription and large amounts of methylation, which lead to the loss of gene expression under most of circumstances [5, 6]. In humans, rDNA sequence is predominantly located on chromosomes 13, 14, 15, 21 and 22 [7]. The region encoding 5S rRNA is primarily situated on chromosome 1 [8]. In mice, these sequences are found on chromosomes 12, 15, 17, 18 and 19 [9, 10], with the region encoding 5S rRNA mainly located on chromosome 8 [11]. Human and mouse sequences encoding rRNAs are highly conserved (percent identity: 5S = 100%, 5.8S = 99%, 18S = 99%, 28S = 85%) [12]. The primary function of rDNA is to form ribosome subunits. rDNA is first transcribed into rRNA in the nucleus by RNA polymerase I (pol I), a rate-limiting factor in ribosomal biogenesis. Additionally, some cell signaling pathways such as MAPK mTOR, MYC, PKC, p53 and RAS/ERK pathways also influence the transcription process. Subsequently, rRNA is processed and modified to assemble with the ribosome protein into the final ribosome subunit. Following ribosomal subunit synthesis, it began to play more prominent roles, such as regulating protein synthesis and transcription factor activity, participating in some post-translational modifications and regulating cell cycle [13, 14]. To meet the different demands of protein synthesis in various tissues under different conditions, multiple modalities such as epigenetic regulation are served to trigger the activation or silencing of rDNA transcription [15–17].
Fig. 1. The structure of rDNA in human cells
DNA methylation usually refers to a modification formed by the addition of methyl groups to the 5’C position of cytosine by DNA methyltransferase (DNMT) [18, 19]. Serving as an essential epigenetic regulator, it plays roles in various biological processes. Relevant research and data have been integrated into epigenetic databases to facilitate researchers’ understanding of advancements [20–22]. rDNA, as one of the key regions of methylation in the human genome, has at least 1500 CpGs on its transcription unit [23]. The methylation levels of rDNA vary across different tissues. A study reported the methylation levels of rDNA promoters in various tissues of adult mice as the following: liver 34%, spleen 53%, brain 48%, testis 25%, and ovary 30% [24]. As far back to 2001, study in mice had clearly identified that methylated rDNA copies are transcriptionally silent [17]. Methylation-mediated silencing of rDNA transcription can contribute to various biological processes such as organismal development, aging, and disease occurrence.
However, due to the high repeatability, near-mitochondrial characteristics and substantial size of rDNA, a lot of relevant information was lost during analysis of next-generation sequencing data, leading to the exclusion of rDNA sequence from the reference genome for a long time. Given this circumstance, research focusing on rDNA methylation is relatively constrained, with even fewer comprehensive reviews on the subject [5, 25]. In this review, we comprehensively summarize the current status and potential applications of research on the relationship between rDNA methylation and embryonic development, aging, and diseases across different species and tissues (Table 1). Additionally, we will provide insights into future directions for rDNA research.
Table 1. Correlation between rDNA methylation and embryonic development, aging, and diseasesTrait typeTraitTissueSpeciesrDNA regionMethylation state changesOther related changesReferenceDevelopmentEmbryonic developmentSperm, germinal vesicle oocytes, metaphase II oocytes, zygote and blastocystsMousePromoterHypermethylation compared to adult tissues [24]SpermMousePromoter, transcribed region and IGSHypermethylation in aged sperm(Peri)Centromeric minor and major satellite DNA, and interspersed LINE1 T repeats hypermethylation in aged sperm [26]RatTranscribed regionHypermethylation in aged sperm- [27]BullTranscribed regionHypermethylation in aged sperm- [26]MarmosetTranscribed regionSlight hypermethylation in aged sperm- [26]HumanPromoter and transcribed regionHypermethylation in aged sperm- [26]OocyteMousePromoter and transcribed regionHypermethylation in aged oocytes- [28]HumanPromoterHypermethylation in oocytes from aging ovarianMethylation changes between individual oocytes from the same donor are less than 5% in most samples [28]ZebrafishTranscribed regionHypermethylationGermline amplification [29]Embryonic day 4.5MousePromoterEmbryo methylation lower than extra-embryo- [24]Gonads and Liver cells of embryonic day 13.5 and 18.5MousePromoterHypermethylation compared to primordial germ cells- [24]Liver cells of embryonic day 18.5MousePromoterHypermethylation compared to embryonic day 13.5- [24]AgingAgingBloodRatPromoterHypermethylation- [14]HumanPromoterNo significant change- [14]Bone marrow cellsMousePromoterHypermethylationThe number of rDNA copies increased and the levels of pre-rRNA transcripts decreased [30]FibroblastsHumanPromoterHypermethylation- [31]Skeleton muscleMousePromoter15 sites were hypomethylated and 345 sites were hypermethylated- [32]HeartRatPromoterHypermethylation- [14]KidneyRatPromoterHypermethylation- [14]LiverRatPromoterHypermethylation- [14]Transcribed regionHypermethylation- [27]Promoter and transcribed regionHypermethylation- [33]HumanPromoter and transcribed regionHypermethylation- [33]DiseasesNeurological diseasesAlzheimer’s disease (AD)Cerebral cortexHumanPromoterHypermethylationAn increase in overall rDNA content [34]Mild cognitive impairment (MCI)Cerebral cortexHumanPromoterHypermethylationAn increase in overall rDNA content [34]Schizophrenia (SCZ)Neuronal and oligodendrocytesHumanTranscribed regionNo significant changeNo significant change in rDNA copy number [35]Autism spectrum disorders (ASD)BrainHumanTranscribed regionNo significant changeNo significant change in rDNA copy number [35]Hematologic diseasesMyelodysplastic syndrome (MDS)CD34 + cellsHumanPromoterHypermethylationDecreased expression of rRNA, and alternation of ribosomal biogenesis [36]Genetic disordersWerner’s syndrome (WS)FibroblastsHumanPromoter, transcribed region and IGSHypermethylationThe steady-state levels of 28S rRNA remained constant over the life span of both normal and WS fibroblasts [37]Down syndrome (DS)Whole bloodHumanPromoterHypermethylationThe number of rDNA copies increased [38]Gender-specific cancersBreast cancerBreastHumanTranscribed regionHypermethylationIncreased methylation is associated with inhibition of estrogen receptor expression [39]BreastHumanPromoter and transcribed regionHypermethylationLower levels of rDNA methylation demonstrated significantly poorer rates of disease-free survival and overall survival [40]Ovarian cancerOvaryHumanTranscribed regionHypermethylationOverexpression [41]Cervical cancerCervical intraepithelial neoplasia tissueHumanPromoterHypomethylationDecondensation of rDNA, and rRNA overexpression [42]Endometrial carcinomaEndometriumHumanTranscribed regionHypermethylationMorphological changes in the nucleoli, ac-companied by overexpression of rRNA [43]Prostate cancerProstateHumanTranscribed regionNo significant changerRNA overexpression [44]ProstateHumanIGSHypomethylationIncreased variation [45]Other types of cancersOral squamous cell carcinoma (OSCC)OralHumanPromoter and transcribed regionNo significant changeDecreased expression of rRNA [46]Esophageal cancerEsophagusHumanIGSHypomethylationIncreased variation [45]Hepatocellular carcinomaLiver, cfDNAHumanIGSHypomethylationIncreased variation [45]Colorectal cancerColorectum, cfDNAHumanIGSPartial tumor samples show hypomethylation- [45]Lung cancerLung, cfDNAHumanIGSPartial tumor samples show hypomethylation- [45]
rDNA methylation and embryonic development
It is widely recognized that during mammalian embryonic development, DNA methylation undergoes dynamic changes, including significant genome-wide “erasure-reconstruction” processes, with the exception of parental imprinting [47–49]. However, research examining the correlation between rDNA methylation and embryonic development stage is notably deficient in systematic approach. It predominantly concentrates on isolated developmental processes in mice, supplemented by studies concerning reproductive cells across various species.
The formation and maturation of germ cells are prerequisites for embryonic development. A study has reported that the variation in rDNA methylation among oocytes derived from the same individual in the majority of humans is less than 5% [28]. In a study conducted on mice, the promoter regions of rDNA in sperm (5.9%), germinal vesicle oocytes (5.4%), metaphase II oocytes (3.2%), zygotes (2.6%) and blastocysts (1.9%) exhibited significantly lower methylated levels compared to those of adult tissues, such as testis (25%), ovary (30%), liver (34%), brain (48%), spleen (53%) [24]. Several studies have identified an increase in rDNA methylation in germ cells with advancing age across various species. It has been discovered that in human sperm, the methylation levels of rDNA transcription units (including UCE, core promoter, 18S rDNA, and 28S rDNA) increase with age, similar to α-satellite DNA and LINE1. Similarly, methylation levels of rDNA in mouse sperm (including intergenic spacer, gene promoter, 18S, and 28S) also increase with age. Additionally, in bovine and marmoset sperm, methylation levels of rDNA (18S and 28S rDNA) rise with individual age, with marmosets showing a slight increase in magnitude [26]. Subsequent research suggested that human ovarian aging is associated with increasing oocyte methylation of the rDNA promoter and UCE, while a weak correlation of oocyte methylation with rDNA promoter, UCE, and 28S rDNA was found in mice [28].
After the mouse zygote undergoes cleavage to form a blastocyst, between embryonic day 4.5 (E4.5) and embryonic day 5 (E5), the blastocyst begins to implant into the uterine wall. After implantation, de novo methylation of rDNA promoters occurs, with the overall methylation level in the embryo (13.9%) being lower than that in the extra-embryo (31%) at embryonic day 7.5 (E7.5). Based on this, the rDNA methylation levels of gonads and fetal liver cells were examined at embryonic day 13.5 (E13.5) and embryonic day 18.5 (E18.5), revealing that the levels were higher than those of primordial germ cells at the respective time points. Additionally, liver rDNA promoter methylation increased from 16.3% at E13.5 to 22.2% at E18.5. These results suggest that de novo methylation of the rDNA promoter is inhibited during germ cell development and global methylation occurs only after implantation. It was also speculated that nucleosome remodeling and histone deacetylase (NuRD) might play a role in the maintenance of hypomethylation of rDNA. Furthermore, during the period from conception to weaning, research has shown a linear correlation between growth limitations resulting from protein restriction and alterations in DNA methylation of rDNA in mouse offspring, which persists into adulthood [50]. These findings offer new insights and perspectives for the study of the methylation characteristics of rDNA during development [24]. Some researches have shown that ribosome biogenesis plays a crucial role in maintaining the fate determination of embryonic stem cells and developmental homeostasis of mammalian [51, 52]. We speculate that some pivotal modifications in rDNA methylation during embryonic development may regulate rDNA transcription and silencing, thereby tightly controlling ribosome biogenesis and promoting the progression of embryonic development at appropriate stages. The Lower levels of rDNA methylation observed at the zygote and blastocyst stages may allow for extensive rDNA transcription, facilitating abundant synthesis of ribosomal RNA essential for the continued differentiation and development of embryonic cells. Therefore, we contend that studying rDNA methylation in embryonic development will contribute to a deeper understanding of the molecular mechanisms of embryonic development and have potential implications for the understanding of the pathogenesis of developmental diseases and applications in embryonic stem cell research.
Apart from mammals, the rDNA of the model organism zebrafish has also been studied during development. Research has shown that zebrafish maintain global methylation levels during sex differentiation without any stage similar to genome-wide methylation erasure in mammals. A specific type of rDNA located in the major sex-linked locus in oocytes (referred to as fem-rDNA) has been identified. Fem-rDNA amplification and demethylation during early development of zebrafish suggest that fem-rDNA amplification is implicated in sex determination, helping to explain the non-Mendelian sex ratio in zebrafish [29].
rDNA methylation and aging
rDNA methylation can be considered a more effective biomarker of biological aging compared to other genomic components due to its comparable accuracy, more concentrated sites, and evolutionarily conserved characteristics [23, 53]. The theory proposed in 2008 suggested that the instability of the rDNA triggers aging and shortens individuals’ life span, which is called “rDNA theory of aging” [53, 54]. Many studies have explored the relationship between rDNA methylation and aging across different species and tissues, with the majority of findings indicating site-specific or overall hypermethylation of rDNA triggered by aging. In blood, elevated rDNA methylation and decreased rRNA levels were reported later in life in rats, although a comparable increase was not conclusively observed in humans [14]. The author further conducted a 9-year longitudinal study on the quality of life in individuals aged 60 to 89 years, revealing a negative correlation between rDNA methylation levels and cognitive abilities, as well as survival chances in the elderly population [14]. In bone marrow, it was observed that with increasing age, there is an elevation in the methylation level and copy number of rDNA, accompanied by a decrease in the levels of pre-rRNA transcripts using 2-year-old and 4-week-old mice [30]. For human fibroblasts, a 2018 report demonstrated an increase in rDNA methylation during in vitro aging [31]. For skeleton muscle, 345 CpG sites in rDNA were found hypermethylated in aged muscle compared to young muscle of mouse, while the number of hypomethylated CpG sites was only 15 [32]. In the heart and kidney of rats, with aging, the methylation of some CpG units in the promoter region increases [14]. For the liver, several studies have reported an age-related increase in DNA methylation at multiple regions of rDNA [14, 27, 33]. Methylation levels of three regions of the rDNA gene locus (promoter, 5’ of 18S, and 5’ of 28S sequences) were measured in young (two months), adult (10 months), and old (18 months) rats, revealing an age-associated increase in DNA methylation [33]. Additionally, a similar increase in methylation occurring in human liver cells was documented for the first time [33].
Based on the age-related changes in rDNA methylation described above, several age prediction models have been developed, utilizing the conservative characteristics of rDNA methylation to assist in the study of aging process. One model has been developed to predict individual’s age based on 15 CpGs within the rDNA transcription unit of human sperm. This model utilizes a relatively small amount of sample size of approximately10 sperm cells, with a prediction error of less than 3 years [26]. Furthermore, the effectiveness of rDNA methylation sites as a clock marker to predict the age of an individual has been tested across multiple distant species, including humans, mice, and dogs. The performance of rDNA is significantly better than that of clocks constructed with other genomic CpG sites as markers, and it is considered to be a reliable predictor of age due to its conserved performance across different species [23]. Among approximately 13,000 base pairs of rDNA sequence in mice, researchers identified 620 CpG sites (66.8%) that provide information about age. Interestingly, many of these CpG sites are conserved across distantly related species; for instance, over 70% of human CpG sites in the 18S and 5.8S genes of rDNA can be found in species as diverse as zebrafish. The elastic network regression model, constructed using methylation levels of 46 rDNA loci, has achieved accurate and precise estimation of the age of individual European lobsters, which will hopefully have economic and ecological value for fisheries management [55]. Furthermore, combining the rDNA methylation clock with genome-wide methylation clocks identified across the entire genome could potentially enhance the accuracy of age prediction [56–59].
Various hypotheses have been proposed regarding the potential cell biological mechanisms through which rDNA methylation may be involved in aging, including nucleolar stress, regulation of RNA polymerase affecting transcription rate, among others. A replication fork barrier (RFB) has been identified within human 47S rDNA, with essential elements including the Sal box and transcription termination factor-1 (TTF-1). Interestingly, it is active only in the hypomethylated rDNA copy, and its mechanism of action is to cooperate with TTF-1 to terminate RNA pol I’s transcriptional activity. The different methylation status of rDNA copies, therefore, could lead to genomic instability, potentially contributing to the development of cancer [60]. Besides, impaired ribosomal biosynthesis due to DNA methylation can also lead to nucleolar stress, which is involved in the pathogenesis of aging and many related diseases [26].
rDNA methylation and diseases
The consequences of impaired ribosomal biosynthesis resulting from rDNA methylation reach far beyond the scope of aging, extending into the intricate landscape of diseases. The resulting nucleolar stress, linked to the pathogenesis of aging, serves as a pivotal link connecting rDNA methylation to a multitude of diseases. This connection prompts an exploration into the specific nuances of how alterations in ribosomal DNA methylation may contribute to the onset and progression of various diseases, thereby unraveling a deeper understanding of the complex interplay between epigenetic modifications and pathological processes.
rDNA methylation and neurological diseases
Decreased rRNA levels and reduced ribosomal activity have been linked to the pathological progression of Alzheimer’s disease (AD) [61–63]. Separate studies have indicated elevated rDNA methylation and increased rDNA content in the cerebral cortex and cerebellum regions of AD patients compared to age-matched controls [34, 64]. Similarly, increased rDNA methylation has been observed in the cortical region of patients with mild cognitive impairment (MCI), an early stage of AD. In the early stages of AD, there is an increase in rDNA methylation, whereas in the late stages, there is an enrichment of low-methylated rDNA units. In contrast, patients with MCI exhibit a broader range of global methylation, with higher methylation levels compared to late-stage AD patients [34]. For another neurological disease, autism spectrum disorder (ASD), a comparative analysis involving 16 ASD samples and 11 control samples did not reveal any significant alterations in rDNA copy number or DNA methylation in the brains of individuals with ASD [35]. Similarly, using 53 neuronal and 42 oligodendrocyte samples, no changes in rDNA methylation or copy number associated with schizophrenia (SCZ) were observed [35].
rDNA methylation and hematologic diseases
With the increase of age, the activity and function of the hematopoietic system decline, which is manifested by the decrease of the number of lymphocytes and the weakening of hematopoietic stem cells (HSC) differentiation function. In CD34 + cells from myelodysplastic syndrome (MDS) patients, there is increased methylation of rDNA promoters, leading to reduced expression of rRNA and disruption of ribosomal biogenesis compared to healthy controls. It is also explored the hypothesis that hypomethylating agents induced an increase in intracellular rRNA transcription, thereby reducing apoptosis, and obtained support from recent data [36].
rDNA methylation and genetic disorders
Werner’s syndrome (WS), also known as progeria short stature disease or adult progeria syndrome, is characterized by premature aging of the skin, a senile appearance, short stature, degeneration of the cardiovascular system, and a higher risk of diabetes and malignant cancer. In vitro studies examining rDNA in fibroblasts from both young and old normal individuals, as well as WS patients, revealed significant hypermethylation in samples from normal elderly individuals and WS patients. This hypermethylation leads to decreased expression of RNA pol II-transcribed genes. Moreover, the life span of fibroblasts in WS patients is significantly shortened [37].
Down syndrome (DS), also known as trisomy 21, is the most common viable chromosomal aneuploidy. The characteristic features of this condition include cognitive impairments, craniofacial anomalies, and early infantile hypotonia [65]. In a study utilizing targeted deep sequencing to assess DNA methylation in the rDNA of 69 adult individuals with DS and 95 controls using whole blood samples, elevated methylation level were identified in the promoter region of rDNA in individuals with DS compared to controls [38]. It has been reported that a significant positive correlation between 45S rDNA methylation and relative rDNA copy number in multiple tissues [35]. Trisomy of chromosome 21 results in most Down syndrome patients having an additional 30–40 copies of the rDNA unit. The increased rDNA methylation leads to silencing of these additional rDNA copies. It may be a dosage compensation mechanism, which ensures the homoeostatic regulation of ribosome biogenesis.
rDNA methylation and cancers
rDNA methylation and sex hormone-regulated cancers
Breast cancer, ovarian cancer, cervical cancer and endometrial cancer are among the major malignancies that adversely affect women’s health worldwide. As early as 2000, it was reported that the average percentage of rDNA methylation in breast tumors was notably higher compared to normal control samples. This increase was associated with the inhibition of estrogen receptor expression [39]. In 2014, further validation of this phenomenon revealed increased methylation at the promoter, 18S, and 28S regions of rDNA. Additionally, DNA methylation levels of several CpG sites within the rDNA locus were found to correlate with the nuclear grade and nucleolar size of tumor tissues [40]. Research on ovarian cancer tumor samples indicated significantly higher methylation levels of 18S and 28S rDNA compared to normal ovarian surface epithelial samples. Furthermore, this hypermethylation could predict the progression-free survival of ovarian cancer patients. Treatment with the demethylating drug 5’-aza-2’-deoxycytidine showed that the rDNA hypermethylation in ovarian cancer led to gene silencing [41]. Investigation into the role of rDNA methylation status and rRNA expression level in cervical intraepithelial neoplasia (CIN) specimens revealed rRNA overexpression in cervical cancer cells, accompanied by demethylation of rDNA promoter and decondensation of rDNA, presumably promoting protein synthesis to support cancer cell proliferation and expansion. Although reports on the multistage development of the disease are still lacking, this study suggested that demethylation of rDNA promoter in cervical cancer cells leads to increased rRNA expression [42]. Analysis of rDNA methylation in 215 endometrial tumors showed that the majority of the tumors exhibited elevated levels of rDNA methylation. Patients with lower levels of rDNA methylation demonstrated significantly worse disease-free survival and overall survival [43].
Prostate cancer ranks as the second most prevalent malignancy among men globally, trailing only lung cancer [66]. Research conducted on human prostate cancer cell lines and clinical samples unveiled no significant correlation between rDNA promoter methylation and rRNA overexpression. Furthermore, it indicated that MYC governs rRNA levels in human prostate cancer, implying a molecular mechanism potentially contributing to nucleolar alterations [44]. However, a recent study suggests that prostate cancer demonstrates reduced methylation in the IGS region of rDNA and heightened variability of rDNA compared to normal tissues [45].
rDNA methylation and other types of cancers
In addition to sex hormone-regulated cancers, variability in rDNA methylation has also been noted in oral squamous cell carcinoma, esophageal cancer, hepatocellular carcinoma, lung cancer, and colorectal cancer. In oral squamous cell carcinoma, rRNA expression is suppressed compared to normal tissues, but no significant DNA methylation changes were observed in the rDNA promoter region [46]. Representative CpG sites of rDNA exhibit a significant decrease in average methylation levels and an increase in variability in esophageal cancer and hepatocellular carcinoma. Partial tumor samples from colorectal cancer, liver cancer, and lung cancer also displayed hypomethylation. Furthermore, the methylation profile of rDNA in cell-free DNA from plasma can serve as a biomarker for the detection of various cancers, including colorectal cancer, lung cancer, and liver cancer [45].
To summarize, dysregulated rDNA methylation is a common feature observed in a range of diseases, including neurodegenerative diseases, hematologic diseases, genetic disorders, and cancers. In ovarian and endometrial cancers, rDNA methylation levels can predict progression-free survival, while rDNA methylation in cell-free DNA from plasma can serve as a biomarker for detecting multiple cancers. Given the extensive remodeling of the epigenome in diseases, changes in rDNA methylation are perhaps unsurprising. However, the key lies in identifying specific rDNA methylation changes that truly indicate disease progression and significantly impact disease development. This will provide valuable insights into disease mechanisms, facilitate the search for early diagnostic markers for diseases, and support therapeutic interventions.
Conclusions
This review provides a comprehensive overview of advancements in research on rDNA methylation concerning embryonic development, age and diseases associations across multiple species. During the preparatory stages of embryonic development, germ cells exhibit lower rDNA methylation compared to somatic cells. Following embryo implantation, there is a remethylation of the rDNA promoter, leading to overall higher methylation levels within the embryo than outside. As organisms age, rDNA methylation increases in the sperm and oocyte of humans, rats, and mice. Various tissues across species, including liver, heart, bone marrow, and skeletal muscle, display heightened rDNA promoter methylation. Changes in rDNA methylation levels are also observed in various types of diseases. rDNA methylation serves not only as an epigenetic clock across multiple species but also as a biomarker for the detection and prognosis of various diseases.
The genetic identity of rDNA units plays a crucial role in determining their epigenetic state. Genetic variations within the rDNA sequence can lead to the emergence of different epialleles, which in turn affect rDNA methylation patterns [67]. For example, specific genetic variants may predispose rDNA units to increased methylation, serving as a mechanism to silence additional rDNA copies. During development, aging, and disease, alterations in rDNA methylation may be influenced by genetic variations associated with rDNA [67], and concurrently coordinated with changes in rDNA copy number [35], thereby potentially regulating rDNA transcription, resulting in an overall modification in protein synthesis rates in the organism. Dysregulation of cellular protein synthesis may significantly contribute to the gradual deterioration of cellular functions associated with aging and diseases.
Despite these advancements, there remains a noticeable gap in research concerning the dynamic patterns of rDNA methylation throughout the entire embryonic development process across various species, as well as its systemic evolution. Questions persist regarding whether rDNA methylation influences processes such as cell differentiation, orientation, and proliferation. Additionally, the precise causal relationship between rDNA methylation and disease or aging process remains elusive. One of the challenges hindering rDNA research is that complete rDNA sequences are typically absent from most standard genome assemblies. The highly repetitive nature of rDNA makes it difficult to accurately analyze and locate rDNA within genome sequences [16, 68]. Additionally, rDNA copy number variation and the characteristic dense tangles further complicate the accurate assignment of reads from sequencing data, as well as the precise identification and quantification of these regions [69].
To advance our comprehension of rDNA methylation, researchers have focused on assembling the rDNA sequence and structure in the human genome. They have employed a specific method to separate the rDNA methylated region from whole-genome bisulfite sequencing (WGBS) data [45]. Additionally, by utilizing the Arachne assembler method, researchers have enhanced the suitability of the reference genome for rDNA localization, enabling precise insertion of rDNA tandem repeat sequences [70]. Recently, researchers have also customized five human and mouse reference genome assemblies, incorporating known rDNA sequences, creating annotation files, and proposing a straightforward workflow for researchers to map and visualize rDNA sequences more effectively [68]. Despite these advancements, the method for assembling rDNA sequences has not been widely embraced, resulting in a dearth of database resources displaying rDNA methylation information. Hence, there are plans to incorporate rDNA methylation profiles from various biological states, such as development, aging, and diseases, into our independently developed MethBank database, which contains cross-species single-base resolution DNA methylation data [21]. By identifying more stable and reliable rDNA methylation markers associated with aging, diseases, and development across multiple species, this endeavor aims to provide new directions for disease detection and intervention based on rDNA methylation, as well as clues for understanding the mechanisms underlying rDNA methylation’s involvement in diverse biological processes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Smirnov E, Chmurciakova N, Cmarko D. Hum r DNA cancer Cells, 2021. 10(12).10.3390/cells 10123452 PMC 870012534943960 · doi ↗ · pubmed ↗
- 2Chebrout M et al. Transcription of r RNA in early mouse embryos promotes chromatin reorganization and expression of major satellite repeats. J Cell Sci, 2022. 135(6).10.1242/jcs.25879835048992 · doi ↗ · pubmed ↗
- 3Shao FQ et al. Methylation of 45S ribosomal DNA (r DNA) is associated with cancer and aging in humans International Journal of Genomics, 2021. 2021.10.1155/2021/8818007 PMC 786195633575316 · doi ↗ · pubmed ↗
- 4Watada E et al. Age-dependent ribosomal DNA variations in mice. Mol Cell Biol, 2020. 40(22).10.1128/MCB.00368-20PMC 758887432900821 · doi ↗ · pubmed ↗
- 5Murach KA et al. Late-life exercise mitigates skeletal muscle epigenetic aging. Aging Cell, 2022. 21(1).10.1111/acel.13527 PMC 876101234932867 · doi ↗ · pubmed ↗
- 6Gensous N et al. Aging and caloric restriction modulate the DNA methylation profile of the ribosomal RNA locus in human and rat liver. Nutrients, 2020. 12(2).10.3390/nu 12020277 PMC 707057131973116 · doi ↗ · pubmed ↗
- 7Razzaq A et al. Ribosomal DNA copy number variation is coupled with DNA methylation changes at the 45S rdna locus. Epigenetics, 2023. 18(1).10.1080/15592294.2023.2229203 PMC 1030549037368968 · doi ↗ · pubmed ↗
- 8Bacalini MG et al. The nucleolar size is associated to the methylation status of ribosomal DNA in breast carcinomas. BMC Cancer, 2014. 14.10.1186/1471-2407-14-361PMC 406228324884608 · doi ↗ · pubmed ↗