A model organism pipeline provides insight into the clinical heterogeneity of TARS1 loss-of-function variants
Rebecca Meyer-Schuman, Allison R. Cale, Jennifer A. Pierluissi, Kira E. Jonatzke, Young N. Park, Guy M. Lenk, Stephanie N. Oprescu, Marina A. Grachtchouk, Andrzej A. Dlugosz, Asim A. Beg, Miriam H. Meisler, Anthony Antonellis

TL;DR
This study uses model organisms to explore the effects of TARS1 gene variants, revealing new insights into the range of possible health issues they can cause.
Contribution
The study introduces a hypomorphic TARS1 allele to uncover new recessive phenotypes in model organisms.
Findings
Two TARS1 loss-of-function variants were identified, including a hypomorphic allele (R433H).
A compound heterozygous mouse model showed lung and skin defects similar to patient phenotypes.
The study expands the known clinical heterogeneity of TARS1-related recessive disease.
Abstract
Aminoacyl-tRNA synthetases (ARSs) are ubiquitously expressed, essential enzymes that complete the first step of protein translation: ligation of amino acids to cognate tRNAs. Genes encoding ARSs have been implicated in myriad dominant and recessive phenotypes, the latter often affecting multiple tissues but with frequent involvement of the central and peripheral nervous systems, liver, and lungs. Threonyl-tRNA synthetase (TARS1) encodes the enzyme that ligates threonine to tRNATHR in the cytoplasm. To date, TARS1 variants have been implicated in a recessive brittle hair phenotype. To better understand TARS1-related recessive phenotypes, we engineered three TARS1 missense variants at conserved residues and studied these variants in Saccharomyces cerevisiae and Caenorhabditis elegans models. This revealed two loss-of-function variants, including one hypomorphic allele (R433H). We next…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
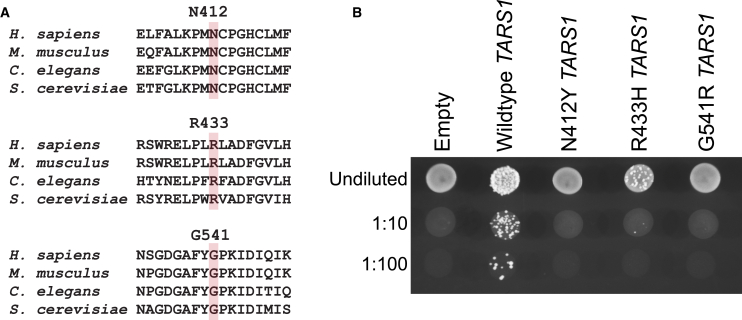 Figure 1
Figure 1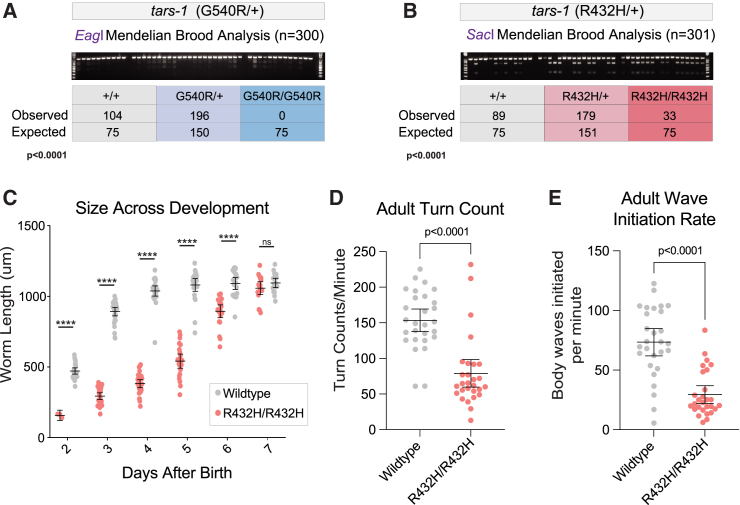 Figure 2
Figure 2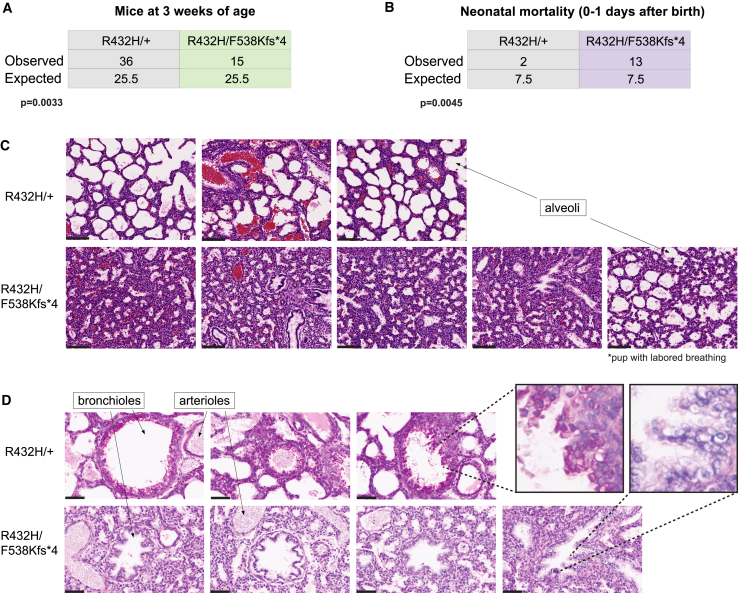 Figure 3
Figure 3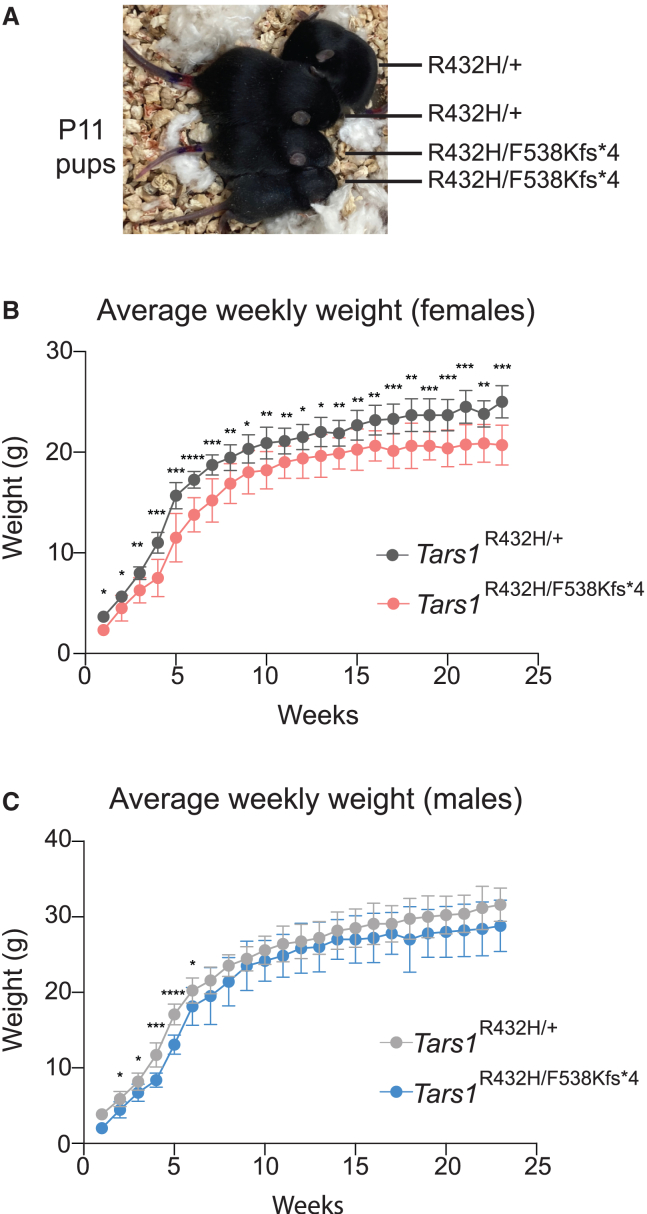 Figure 4
Figure 4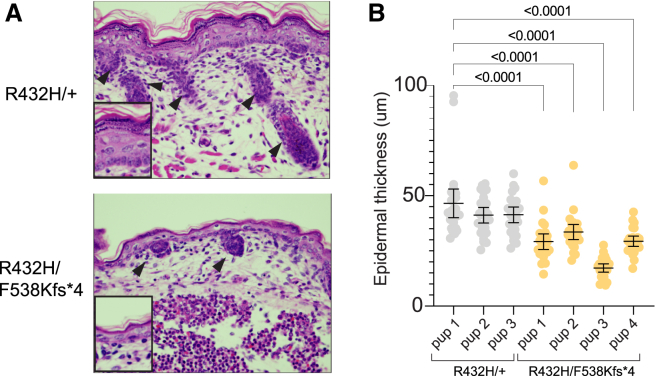 Figure 5
Figure 5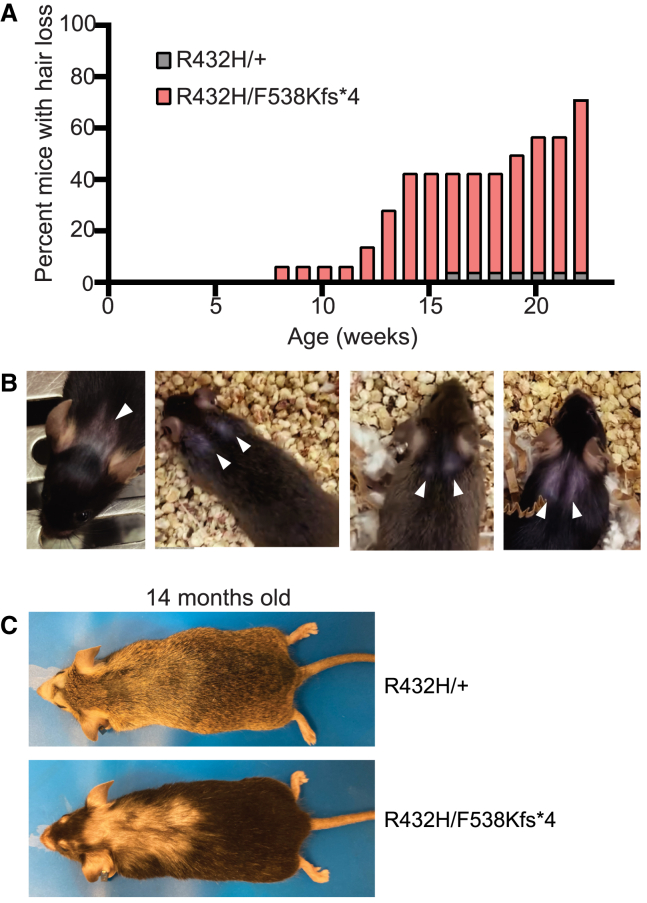 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Rare Diseases · Mitochondrial Function and Pathology · CRISPR and Genetic Engineering
Introduction
Aminoacyl-tRNA synthetases (ARSs) are a family of ubiquitously expressed, essential enzymes that charge tRNA molecules with cognate amino acids, which constitutes the first step of protein translation.1 The human nuclear genome encodes 37 ARS loci, with 17 encoding mitochondria-specific enzymes, 18 encoding cytoplasm-specific enzymes, and two encoding enzymes that function in both compartments.2^,^3 Variants in genes encoding ARSs have been implicated in a spectrum of genetic diseases with all 37 loci implicated in recessive multisystem disorders.4^,^5^,^6 These disorders are caused by bi-allelic variants that severely impair gene function but do not eliminate it, as total loss of any ARS is incompatible with life. Bi-allelic pathogenic variants that affect mitochondrial ARSs tend to cause phenotypes in tissues with a high metabolic demand, including leukoencephalopathies,7^,^8 myopathies,9 and liver disease.10^,^11 Bi-allelic pathogenic variants in ARS genes encoding cytoplasmic enzymes often affect a wider array of tissues but typically include a neurological component. The recessive neurological phenotypes associated with cytoplasmic ARSs include hypomyelination,12^,^13 microcephaly,14^,^15 seizures,16^,^17 sensorineural hearing loss,18^,^19 and developmental delay.20^,^21^,^22 Interestingly, variants in some ARS loci cause tissue-restricted or tissue-predominant recessive phenotypes.23 For example, although variants in FARSA,24 FARSB,25^,^26 IARS1,22 MARS1,27 and YARS128 frequently cause liver dysfunction as one component of a multisystem disease, LARS1 variants are the most consistent cause of a severe, acute form of infantile liver failure.29^,^30^,^31 Similarly, pulmonary disease is pronounced in individuals with bi-allelic FARSB25^,^26^,^32 and MARS1 variants,27^,^33 including a MARS1-specific form of pulmonary alveolar proteinosis.34 The clinical and mechanistic heterogeneities of recessive ARS-related diseases are poorly defined; advancing our knowledge in this area will require generating and characterizing relevant animal models.35
Due to the conservation of ARS genes across evolutionarily diverse species, multiple model organisms can be used to study ARS biology and to investigate the impact of pathogenic variants. These models include yeast, worms, fruit flies, and zebrafish.36^,^37 Mammalian models have historically been limited to studying forms of ARS-mediated dominant peripheral neuropathy38^,^39^,^40 but are increasingly employed for modeling ARS-mediated recessive diseases, including models for Dars1,41^,^42 Iars143, Fars2,44 Sars2,45 and Wars2.46 Moving forward, mouse models will be critical tools to understand why certain tissues are particularly sensitive to loss-of-function variants in specific ARS genes, as these questions must be addressed in a model organism with relevant tissue types.
To build a relevant model system pipeline of an understudied ARS gene, we focused on threonyl-tRNA synthetase (TARS1). When this study began, TARS1 had not been implicated in any human disease phenotype. Bi-allelic loss-of-function TARS1 variants have since been reported in two patients with a recessive brittle hair phenotype.47 Additionally, bi-allelic TARS1 variants were identified in an individual with cerebral palsy and developmental delay, although these variants have not yet been functionally evaluated.48 To obtain a more complete assessment of TARS1-related recessive phenotypes in a manner that is not limited by patient ascertainment, we generated a model organism pipeline comprising yeast, worm, and mouse. We first engineered three TARS1 missense variants predicted to cause a loss-of-function effect and tested for these effects in yeast and worm models, which revealed one of these variants (R433H) as a hypomorphic allele. We then used R433H to study the effects of partial loss of TARS1 function in a compound heterozygous mouse model (R432H/null; R432 is the orthologous position of R433 in mouse Tars1). This model presents with some phenotypes that are reminiscent of the trichothiodystrophy recently attributed to bi-allelic TARS1 variants.47 This model also presents with distinct lung and skin phenotypes not previously associated with TARS1. In sum, this study expands the potential clinical heterogeneity of TARS1-related recessive disease, which should guide future clinical and genetic evaluations of relevant patient populations.
Results
Identification of three loss-of-function TARS1 variants
The model organism Saccharomyces cerevisiae, or Baker’s yeast, provides a tractable eukaryotic system for studying highly conserved human genes, as well as disease-associated variants that affect the function of these genes.49^,^50 For aminoacyl-tRNA synthetase (ARS) genes, S. cerevisiae has been a reliable model to study pathogenic variants and test them for loss-of-function effects.37 Because ARS genes are highly conserved across evolution, the human open reading frame can often complement loss of the endogenous S. cerevisiae ortholog, resulting in a “humanized” yeast model. In this model, yeast growth is interpreted as a proxy for ARS function, since loss of ARS function will impair cell survival and colony formation.
To design candidate recessive variants in TARS1, we mutated three residues in the TARS1 open reading frame that are highly conserved among human, mouse, worm, and yeast (Figure 1A). These variants—GenBank: NP_001245366.1:p.(N412Y), GenBank: NP_001245366.1:p.(R433H), and GenBank: NP_001245366.1:p.(G541R)—were designed to recapitulate the types of amino-acid changes frequently observed in patient populations. These variants were also designed in the aminoacylation domain of the protein to increase the chance that they would disrupt the protein’s catalytic function. To assess whether these variants affect human TARS1 function, a complementation assay was performed using an S. cerevisiae strain with the endogenous THS1 deleted. Yeast viability was maintained with a pRS316 vector51 expressing the yeast THS1, along with URA3. The pYY1 vector52 expressing either wild-type or mutant human TARS1 was transformed into yeast, then yeast were plated on 5-FOA, which selects for the loss of the maintenance vector expressing URA3 and THS1.53 Wild-type TARS1 supported yeast growth, demonstrating that human TARS1 can function in yeast (Figure 1B). Transformation with N412Y TARS1 or G541R TARS1 did not lead to formation of colonies, indicating that these two variants significantly impair TARS1 function. Transformation with R433H TARS1 did support some yeast growth but caused significantly reduced colony formation compared to wild-type TARS1 (Figure 1B), indicating partial impairment of TARS1 function (i.e., a hypomorphic allele).Figure 1. Engineered TARS1 variants display a loss-of-function effect in S. cerevisiae(A) Conservation analysis of N412, R433, and G541 TARS1 in H. sapiens (NP_001245366.1), Mus musculus (AAH55371.1), C. elegans (NP_001022033), and S. cerevisiae (NP_116578.3). The targeted residues are highlighted in pink, surrounded by flanking sequences from evolutionarily diverse species.(B) A representative image is shown from three replicates of S. cerevisiae haploid strains with THS1 deleted and transformed with a vector with no TARS1 insert (“Empty”), or with one to express wild-type, N412Y, R433H, or G541R TARS1. Yeast samples were spotted on media containing 5-FOA in serial dilutions (undiluted, 1:10, or 1:100) and then grown at 30°C.
Homozygosity for G540R tars-1 is lethal in worm
To explore how loss-of-function TARS1 variants impact the physiology of a multicellular organism, we modeled two of the above variants—the severe loss-of-function G541R and the partial loss-of-function R433H—in worm (Caenorhabditis elegans). These variants were each introduced into the endogenous worm tars-1 locus using CRISPR-Cas9-mediated gene editing, with synonymous variants introduced in cis to create restriction digest sites to facilitate genotyping (EagI for G540R and SacI for R432H) (Figure S1). Of note, the worm amino acid number differs from the human number by one (Table S1). To minimize effects from possible off-target CRISPR variants, G540R/+ worms were back-crossed to the ancestral N2 strain five times, and R432H/+ worms were back-crossed six times. Then, to assess if either of these variants were grossly deleterious in the homozygous state, heterozygous hermaphrodites were allowed to self-fertilize, and offspring were genotyped at the late larval L4 stage or early P1 adult stage to detect deviation from expected Mendelian ratios. In the case of the G540R/+ hermaphrodites, no G540R/G540R offspring were recovered out of 300 worms, indicating that homozygosity for G540R is lethal (Figure 2A). These data confirm that G540R is a loss-of-function allele, validating the results of the yeast complementation assay (Figure 1B). However, because the G540R/G540R genotype did not produce viable animals for additional phenotypic characterization, this variant was not included in further studies. In contrast, when the offspring of R432H/+ hermaphrodites were genotyped, R432H/R432H homozygotes were identified. Only 33 homozygotes were identified, whereas 75 would be expected if homozygosity for R432H was benign (p < 0.0001; Figure 2B). This indicates that homozygosity for R432H is deleterious but not completely lethal, consistent with the yeast complementation data indicating that R432H is a hypomorphic allele (Figure 1B).Figure 2R432H tars-1 impairs viability and locomotion and delays development in C. elegans(A) Genotype analysis of offspring from five broods of G540R/+ tars-1 hermaphrodites. A representative genotyping gel image is shown. The observed number of each genotype is shown, along with the expected number from Mendelian segregation of a benign variant. A chi-square test between observed and expected numbers was performed to determine statistical significance (p < 0.0001).(B) Genotype analysis of offspring from four broods of R432H/+ hermaphrodites, exactly as described in (A).(C) Measurements of body length of R432H/R432H tars-1 worms and wild-type tars-1 worms at 6 days after birth. On day 2, n = 3 for R432H/R432H, then n = 18–30 worms for each subsequent day. For wild-type worms, n = 18–30 for each day.(D) Turn count per minute for R432H/R432H worms (n = 28) and wild-type worms (n = 28) at adult stage P9.(E) Number of body waves initiated from either the head or the tail per minute, for R432H/R432H worms (n = 28) and wild-type worms (n = 28) at P9. For (C)–(E), bars indicate the mean value and 95% confidence intervals. Statistical significance was evaluated using an unpaired t-test with Welch’s correction; ∗∗∗∗p < 0.000001; ns = not significant.
R432H tars-1 causes recessive developmental delay and locomotion defects in worm
One possible explanation for the depletion of R432H/R432H tars-1 worms in the Mendelian analysis was that this population was under-sampled compared with R432H/+ and wild-type worms. This might occur if developmental delay prevented them from reaching the genotyping time point at the same rate as wild-type or R432H/+ tars-1 worms. To investigate this possibility, a population of R432H/R432H tars-1 worms and wild-type N2 worms were age-synchronized. The physical size of the cohort was tracked for over 7 days. Beginning 48 h after hatching, worm length was measured each day using the WormLab video and software system. R432H/R432H tars-1 worms were consistently smaller than wild-type controls until day 6 or 7 (Figures 2C and S2A). Whereas wild-type worms reach a mature size of approximately 1 mm 3–4 days after birth, R432H/R432H tars-1 worms do not reach this size until 6–7 days after birth.
We next investigated whether R432H might impair locomotion. We performed a thrash assay with adult worms 9 days after they reached adulthood (P9). Here, R432H/R432H tars-1 worms were age-matched to wild-type N2s by synchronizing embryo production. The WormLab video capture and analysis system was used to record 1-min videos of worms swimming in M9 buffer, track their motion, and calculate locomotion parameters. R432H/R432H worms display significant thrash impairment (Figures 2D and 2E; Video S1), indicating that reduced tars-1 function affects the neuronal circuitry or muscular function governing worm locomotion. This locomotion defect was also present in the L4 larval stage, although less pronounced (Figures S2B and S2C). Combined with the significant delay in body size, these data indicate that R432H tars-1 produces significant phenotypes in a multicellular eukaryotic organism.
Video S1. R432H/R432H worms display significant locomotion defectsA 10-s video of adult (P9) wild-type worms (N2 strain) thrashing in liquid M9 media, followed by a 10-s video clip of P9 R432H/R432H tars-1 worms thrashing in liquid M9.
Partial loss of Tars1 function causes neonatal lethality in mouse due to lung defects
To determine how R433H impacts a more complex mammalian system—including defining any specific tissues that might be especially sensitive to partial loss of TARS1 function—we developed a mouse model of R433H TARS1. The R432H variant was introduced into the mouse Tars1 locus using CRISPR-Cas9-mediated gene editing. (Like C. elegans, the mouse amino acid number is 432 [Table S1].) To first determine if R432H caused neonatal lethality in the homozygous state, a Mendelian analysis was performed on the offspring of a Tars1^R432H/+^ heterozygote intercross. Out of 43 genotyped offspring, R432H homozygotes were recovered at a frequency that did not significantly deviate from the predicted 25% (Figure S3A) and were grossly normal throughout adulthood. These observations are consistent with our data from yeast and worm (see above) demonstrating that R432H Tars1 retains partial function. However, it also suggests that, unlike yeast and worm, mice are less sensitive to this degree of Tars1 impairment, and that additional reduction of Tars1 function might be needed to observe a phenotype in a mammalian model. To this end, we generated a mouse Tars1 null allele (F538Kfs∗4) and crossed mice heterozygous for this lesion to Tars1^R432H/R432H^ homozygous mice. The F538Kfs∗4 null allele produces a premature stop codon in exon 14, leading to a reduction in protein levels (Figure S3C) and homozygous lethality (S3B). The R432H/F538Kfs∗4 genotype resembles many individuals with recessive ARS-mediated disease who are compound heterozygous for a hypomorphic missense allele and a null allele.4
To assess the effect of the R432H/F538Kfs∗4 Tars1 genotype on viability, offspring of the cross between Tars1^R432H/R432H^ and Tars1^F538Kfs∗4/+^ mouse strains were genotyped at 3 weeks of age. Out of 51 pups, we only recovered 15 Tars1^R432H/F538Kfs∗4^ (Figure 3A) offspring, indicating decreased viability prior to 3 weeks. An analysis of neonate deaths across four litters showed that pups that died at P0 were enriched for the R432H/F538Kfs∗4 genotype; out of 15 genotyped animals presenting with neonatal death, 13 were R433H/F538Kfs∗4 mice (Figure 3B). To gain insight into the neonatal pathology, a cohort of four P0 Tars1^R432H/F538Kfs∗4^ pups and three age-matched Tars1^R432H/+^ littermates were collected for histology. The four Tars1^R432H/F538Kfs∗4^ pups all died within a few hours of birth. Interestingly, one additional Tars1^R432H/F538Kfs∗4^ pup was found immediately after birth with traces of birth fluids still visible, exhibiting visibly labored breathing and a failure to right itself. This additional pup was included in the cohort to assess for a respiratory phenotype. All pups were fixed in formalin overnight and washed with 70% ethanol. Subsequently, sagittal sections were prepared and stained with H&E to detect gross morphological changes, and with periodic acid-Schiff (PAS) staining to detect changes in glycoproteins and mucins.Figure 3. Depleted TARS1 function causes reduced viability and a lung phenotype in a mouse model(A) Genotype analysis of Tars1^R432H/R432H^ and Tars1^F538Kfs∗4/+^ offspring, genotyped upon weaning at 3 weeks of age. The observed and expected number of each genotype is shown.(B) Genotype analysis of 15 deceased pups, identified within 1 day after birth. The observed and expected number of each genotype is shown. For (A) and (B), a chi-square test was used to determine if the difference between the number of observed and expected genotypes was statistically significant.(C) H&E staining of lung sections from three Tars1^R432H/+^ P0 pups (top row) and five Tars1^R432H/F538Kfs∗4^ P0 pups (bottom row). All Tars1^R432H/+^ pups were alive when identified at P0. The first four Tars1^R432H/F538Kfs∗4^ pups were dead at P0; the fifth was found alive with a gasping, labored breathing pattern. Arrows point to examples of alveoli, which are collapsed in R432H/+ mice. The black scale bar is 100 μm.(D) PAS staining of lung sections from three Tars1^R432H/+^ P0 pups (top row) and four Tars1^R432H/F538Kfs∗4^ P0 pups (bottom row), with labeled examples of bronchioles and arterioles. The inset highlights the magenta PAS signal in the bronchioles of Tars1^R432H/+^ mice, and the absence of PAS signal in the collapsed bronchioles of Tars1^R432H/F538Kfs∗4^ mice. The black scale bar is 50 μm.
The primary finding from H&E staining was an absence of air in the lungs of the four P0 Tars1^R432H/F538Kfs∗4^ mice that died shortly after birth. Whereas the alveoli of Tars1^R432H/+^ control animals were expanded with air, the alveoli of Tars1^R432H/F538Kfs∗^ mice were collapsed (Figure 3C). Considering the otherwise mature body development of these pups, this indicates that they died upon birth or immediately afterward from an inability to breathe. Interestingly, the additional Tars1^R432H/F538Kfs∗4^ pup found alive immediately after birth had only partially expanded alveoli, which correlates with the observed labored breathing. Additionally, while the bronchioles of Tars1^R432H/+^ control mice are replete with the magenta PAS+ signal of secretory cells, this signal is absent from the collapsed bronchioles of Tars1^R432H/F538Kfs∗4^ animals (Figure 3D). To determine if the absent PAS+ signal indicated a loss of these secretory club cells or a significant impairment in their function, we stained similar sections with an antibody against club cell secretory protein (CCSP), an abundant lung protein primarily produced and secreted by the bronchiolar club cells in mouse.54 This revealed no significant difference in CCSP levels between Tars1^R432H/F538Kfs∗4^ mice and control littermates (Figure S4), indicating that these club cells are present and grossly functional. Another possible explanation for the reduction in bronchiolar PAS+ signal was a reduction of specific threonine-rich glycoproteins like mucins, which may be poorly translated in cells with reduced Tars1 function. Previous work in pancreatic cancer cells demonstrated that threonine starvation or knockdown of TARS1 decreases mucin 1 (MUC1) protein levels.55 As MUC1 is also a critical airway protein,56 we stained our P0 pup lung sections with anti-MUC1 to determine whether decreased MUC1 levels were responsible for the decreased PAS+ signal. There was no significant difference between Muc1 signal in Tars1^R432H/F538Kfs∗4^ pups and their littermate controls (Figure S4). Further investigation will be required to determine the underlying mechanism of the loss of PAS+ signal and the pathophysiology of lung dysfunction in mutant Tars1 mice.
Partial loss of Tars1 function causes growth restriction with skin and hair abnormalities
While the studies described here were under way, a report of two patients with bi-allelic TARS1 variants and triochothioydstrophy (TTD) was published.47 The phenotypes described in these two patients included delayed physical development, ichthyosis, and collodion baby, and the brittle hair of TTD. Interestingly, Tars1^R432H/F538Kfs∗4^ mice display phenotypes that are reminiscent of these human disease features. For example, Tars1^R432H/F538Kfs∗4^ mice that survived to adulthood were, on average, smaller than their Tars1^R432H/+^ littermates (Figure 4). Reduced body weight was more consistent in females (Figure 4B) than males (Figure 4C), who reach a normal body size by 7 weeks of age. This reduced size is consistent with the delayed physical development described in TARS1 patients,47 and with the growth restriction phenotypes in patients with other ARS-mediated recessive disease.14^,^22^,^32^,^57Figure 4. Depleted Tars1 function causes reduced body size in a mouse model(A) Image of four littermates at P11, grouped together for comparison of body size. The genotype of each mouse is provided.(B) The average weekly weights of female Tars1^R432H/F538Kfs∗4^ mice (n = 9) and female Tars1^R432H/+^ (n = 11) littermates are shown, until 23 weeks of age.(C) The average weekly weights of male Tars1^R432H/F538Kfs∗4^ mice (n = 6) and male Tars1^R432H/+^ (n = 12) littermates are shown, until 23 weeks of age. For (B) and (C), bars represent the mean value and 1 standard deviation. An unpaired t-test was performed for each week to determine if the difference between the two genotypes was statistically significant. ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05. All values in (C) that are not marked with an asterisk are not significantly different.
We also detected skin and hair abnormalities in the Tars^R432H/F538Kfs∗4^ P0 pups and adult mice. The pups had a thinner epidermal layer than control littermates, with fewer layers of stratum corneum (Figures 5A and 5B). They also displayed variable degrees of hair follicle hypoplasia (Figure 5A). This is unlike the only existing mouse model of TTD, which models a patient mutation in XPD, the most frequently mutated TTD gene.58 In contrast to the Tars1 mice, this TTD model exhibits a thicker epidermal layer. Adult Tars1^R432H/F538Kfs∗4^ mice also displayed a striking postnatal hair phenotype, although it did not resemble the sparse, brittle hair associated with TTD, and was not as severe as the hair loss previously described in the TTD mouse model.58 In a nine-litter cohort, 10 out of 14 Tars1^R432H/F538Kfs∗4^ mice (71.4%) lost hair on their heads and/or upper back by 23 weeks of age, compared with one out of 23 Tars1^R432H/+^ littermates (4.4%). Hair loss onset occurred between 13 and 23 weeks of age (Figure 6A) and followed a stereotypic pattern of bald spots on the head and/or along the scapula of the upper back (Figure 6B). In more advanced stages, it spanned the entire upper back (Figure 6C), although it did not encompass the majority of the body as previously described for TTD mice, nor did it grow back in cycles of loss and regrowth.58 To more thoroughly define this phenotype, histopathology was performed on hair samples from the affected regions for three Tars1^R432H/F538Kfs∗4^ mice and three Tars1^R432H/+^ littermates; one pair was 2 months old, another was 12 months old, and the third was 14 months old. Analysis of H&E staining did not reveal gross abnormalities in hair follicles (data not shown), although this analysis was complicated by the asynchronous hair cycling of adult mice. We also did not observe the classic “tiger tail banding pattern” seen under polarizing microscopy hair from TTD patients.59 Taken together, our data demonstrate that partial loss of TARS1 function causes unusual hair and skin phenotypes in mouse, impairs body weight, and causes a partially penetrant but severe respiratory deficiency.Figure 5. Depleted Tars1 function causes skin phenotypes in a mouse model(A) H&E staining of dorsal skin sections from P0 pups. The upper panel shows a representative image of skin from a Tars1^R432H/+^ mouse, and the bottom panel shows a representative image of skin from a Tars1^R432H/F538Kfs∗4^ mouse. Black arrows point to hair follicles in each image.(B) Measurements of epidermal thickness on four Tars1^R432H/F538Kfs∗4^ P0 pups and three Tars1^R432H/+^ P0 littermates (n = 25 measurements per pup). Bars indicate the mean value and 95% confidence interval. Statistical significance was determined with a one-way ANOVA with Šidák’s multiple comparisons testing, comparing all animals with R432H/+ pup 1. Only p values < 0.05 are shown (differences between R432H/+ pups were not statistically significant).Figure 6. Depletion of Tars1 function causes hair loss in a mouse model(A) The cumulative percentage of Tars1^R432H/F538Kfs∗4^ mice (pink, n = 14) and Tars1^R432H/+^ mice (gray, n = 23) with hair loss on the back of the head or upper back is shown, until 23 weeks of age.(B) Representative images of four individual Tars1^R432H/F538Kfs∗4^ mice with hair loss; white arrows point to the consistent pattern of upper back bald patches. The depicted mice are between 10 weeks and 17 weeks of age.(C) A representative image of hair phenotypes in Tars1^R432H/+^ (top) and Tars1^R432H/F538Kfs∗4^ (bottom) animals. Note that extended hair loss stretches from the head to the middle of the back in the Tars1^R432H/F538Kfs∗4^ mouse, at 14 months of age. A Tars1^R432H/+^ littermate is shown above, with signs of barbering by the nose and mild age-related hair thinning on the back.
Discussion
In this study, we leveraged the established characteristics of pathogenic ARS variants to develop a model system pipeline for predicting the clinical heterogeneity of TARS1-related recessive disease. We successfully engineered two loss-of-function TARS1 missense variants, including a partial loss-of-function allele, R433H, that we employed to explore recessive phenotypes across three model organisms. This analysis revealed that R433H: (1) partially reduced yeast growth in yeast complementation assays; (2) caused developmental delay and locomotion defects in homozygous worms; and (3) caused lung failure, decreased body size, and skin and hair defects when modeled in trans with a null allele in mice. While skin and hair phenotypes are seen in our mouse mutants, they do not precisely mimic those recently described for humans with TARS1 variants. The remaining phenotypes are unique and have not been previously described. While there may be many reasons for this, one possible explanation is that while the well-characterized human missense variants lie within the N2 editing domain, R433H lies within the aminoacylation domain.47^,^60 This raises the possibility that there are subtle differences in how these variants impact cellular physiology. Overall, our data indicate that phenotypic heterogeneity will ultimately be observed in human TARS1-related recessive disease, and that individuals with bi-allelic TARS1 variants should be carefully evaluated for lung disease.
It is interesting to consider why some tissues may be particularly sensitive to reductions in TARS1 function. One possibility is that critical proteins with a particularly high threonine content, such as mucins, are more dramatically affected by decreased Tars1 activity. This could lead to defects in the tissues that rely heavily on these proteins, such as the lung. Interestingly, the gut is also dependent on mucin synthesis.61 Although preliminary investigation of gut histology in P0 mice did not identify any changes in PAS signal, careful assessments for gut phenotypes should be performed on patients who are homozygous or compound heterozygous for pathogenic TARS1 variants. Another possibility is that decreased Tars1 activity reduces the available population of charged tRNA^Thr^, triggering eIF2α phosphorylation, which then leads to decreased global protein translation. This might affect cells with a high demand for protein translation, such as transient-amplifying progeny of stem cells. For example, if aging hair follicle stem cell progeny cannot properly translate the large mass of proteins required for differentiation of the multiple cell types comprising the mature hair follicle and hair shaft, this could explain a failure to regrow hair in the adult Tars1^R432H/F538Kfs∗4^.
In summary, this study demonstrates the efficacy of using variant engineering and a tiered model organism approach to predict the pathogenicity of variants in ARS genes. While additional research on human subjects and animal models will be required to fully define the clinical heterogeneity of TARS1-related disease, the data presented here should be useful in genetic and clinical evaluation of relevant patient populations.
Materials and methods
Generation of TARS1 expression constructs
The open reading frame (ORF) of the TARS1 transcript (GenBank: NM_001258437) was amplified from HeLa cell cDNA, using primers with the attB1 and attB2 gateway recombination sequences (primer sequences in Table S2). These amplicons were purified with Qiagen Spin Miniprep columns and recombined into pDONR221 using Gateway cloning technology (Invitrogen). The recombination reaction was then transformed into Top10 cells (Invitrogen) to isolate clonal populations. Individual bacterial colonies were selected and grown in media containing kanamycin, which selected for the kanamycin resistance cassette on pDONR221. Plasmids were then isolated using the Qiagen Miniprep kit and genotyped by digesting with BsrGI (New England Biolabs) to detect the presence of the TARS1 insert. Clones with successful insertions were analyzed by Sanger sequencing to ensure absence of variants introduced by amplification errors. To introduce variants into the TARS1 ORF, site-directed mutagenesis was performed with the QuickChange II XL Site-Directed Mutagenesis Kit (Agilent) (primer sequences Table S2). The reaction was transformed into Top10 cells and grown in Lysogeny broth containing kanamycin to select for pDONR221. Plasmid DNA was isolated and sequenced as above, to ensure successful mutagenesis. Then, the Gateway LR reaction was used to recombine the wild-type or mutant TARS1 into the vector pYY1. This vector has a 2-μm origin of replication, resulting in a high copy number per cell, as well as the ADH1 promoter, resulting in strong constitutive TARS1 expression. Recombinants were transformed into Top10 cells, which were plated on ampicillin to select for the ampicillin resistance cassette on pYY1. Plasmids were extracted, purified, and digested with BsrGI to identify successfully recombined clones.
Yeast complementation assays
Yeast complementation assays were performed with the ΔTHS1 strain (Horizon Discovery, Clone ID 21471). Yeast viability was maintained with a pRS316 vector that expresses wild-type THS1 from the endogenous S. cerevisiae promoter. pRS316 also carries the auxotrophic marker URA3 and has a yeast centromere sequence that results in a low copy number per cell. The pYY1 vector (expressing wild-type TARS1, mutant TARS1, or an empty control) was transformed into yeast with a standard lithium acetate transformation, performed at 30°C with 200 ng of plasmid. Yeasts were grown on solid media without uracil and leucine, which selected for cells with both pRS316 and pYY1. Yeasts were grown for 3 days at 30°C, then individual colonies were picked into 2 mL liquid media lacking uracil and leucine. These cultures were grown for 2 days at 30°C, shaking at 275 rpm. Then, 1 mL of saturated culture was centrifuged at 15,000 rpm for 1 min and cell pellets were re-suspended in 50 μL water. Yeasts were serially diluted to 1:10, 1:100, or 1:1,000 using water. Ten microliters of each dilution (included undiluted yeast) was spotted on complete media containing 5-FOA (Teknova), which selects for cells that have spontaneously lost the pRS315 vector expressing URA3 and THS1.53 After 3 to 5 days, yeast growth was visually inspected.
Generation of the G540R and R432H tars-1 C. elegans strains
To generate the G540R and R432H tars-1 models, CRISPR-Cas9 genome editing was performed according to previously described methods.62 Briefly, the gonadal tract of P1 adult worms was injected with an injection mix of: 300 mM KCl, 20 mM HEPES, 2.5 ng/μL pCFJ90, 50 ng/μL single-stranded oligonucleotide homologous donor repair template (Integrated DNA Technologies), 5 μM single guide (sg) RNA (Synthego), and 5 μM Cas9 protein (Integrated DNA Technologies). Sequences for the repair templates and guide RNAs can be found in Table S3. Injected worms were then placed on single 35-mm plates of nematode growth media (NGM) and fresh OP50 bacteria as a food source. Approximately 2 days after injection, plates were screened for the presence of F1 progeny expressing the pCFJ90 marker, which expresses mCherry in the pharyngeal muscles. This enriches for worms that were exposed to the injection mix, increasing the likelihood of identifying a worm subjected to genome editing. The mCherry-positive F1s were singled to individual plates and allowed to produce their own offspring (F2). Then, the F1 worms were placed in lysis buffer (50 mM KCl, 10 mM Tris-HCl pH 8.3, 2.5 mM MgCl_2_, 0.45% NP-40, 0.45% Tween 20, 1 mg/mL proteinase K) and lysed with incubation at −80°C for 1 h, incubation at 65°C for 1 h, and incubation at 95°C for 15 min. To genotype worms, the targeted tars-1 region was amplified by PCR (primer sequences in Table S2) using Q5 PCR mix (New England Biolabs). Amplicons were then purified with DNA Clean and Concentrator kits (Zymo Research) and digested with the appropriate restriction enzyme (EagI for G540R or SacI for R432H, New England Biolabs). Digested PCR products were separated on a 1% agarose gel and analyzed to identify successful integration of the restriction site. The undigested PCR product from F1s with successful gene editing events was submitted for Sanger sequencing to confirm proper insertion of the restriction site and the desired tars-1 variant. The offspring of these F1 worms were then maintained for subsequent experiments. To reduce possible off-target variants caused by CRISPR-Cas9 editing, R432H/+ tars-1 worms were back-crossed to the ancestral N2 strain six times. To assess the Mendelian ratios of the offspring of R432H/+ tars-1 worms, 6–8 worms from the R432H/+ strain were singled to individual 35-mm plates with OP50, allowed to self-fertilize and produce progeny, then genotyped to confirm heterozygosity for R432H. After confirmation, individual progeny were picked into wells of a 96-well plate for genotyping and Mendelian ratio analysis. This was repeated four times for a total of 301 genotyped offspring.
Measuring worm body size through development
To identify differences in rates of development, R432H/R432H tars-1 worms and wild-type N2 worms were first age-synchronized by placing approximately 25 adult worms on a 60-mm plate with NGM and OP50, letting them produce embryos for 4–5 h, and then removing the adults. After 48 h, worms were transferred to unseeded 35-mm NGM plates in batches of 4–5 worms. These worms were filmed and analyzed using the WormLab System (MBF Biosciences). Plates were filmed for 30-s intervals, with the camera set at 4.81 μm/pixel for R432H/R432H worms (Setting 1 on the WormLab camera apparatus [MBF Bioscience]) and 8.47 μm/pixel for N2 worms (Setting 3 on the WormLab camera apparatus [MBF Bioscience]). After filming, worms were moved to new NGM plates seeded with OP50. Filming was repeated every 24 h up to 168 h, or 7 days, after birth (as R432H/R432H worms increased in size, filming was performed with the camera setting at 8.47 μm/pixel). All videos were analyzed with the WormLab software (MBF Bioscience), and the “worm length” parameter was extracted to compare the size of R432H/R432H tars-1 worms and N2 worms over the course of development.
Worm thrash assays
Thrash assays were performed to detect changes in worm movement. The bottom of each well of a Nunc 4-well dish (Thermo Scientific) was coated with 2.5% agarose. An amount of 500 μL liquid M9 media (22 mM H_2_KO_4_P, 42 mM HNa_2_O_4_P, 85 mM NaCl, 1 mM MgSO_4_) was added to each well, and 1–5 worms (wild-type or R432H/R432H tars-1) were placed in the M9. Worms were allowed to acclimate for 30–60 s before they were filmed with the WormLab System (MBF Biosciences) for 1 min. Only worms with at least 1,000 frames or 30 s of high-quality video were included in subsequent analysis. To identify defects in locomotion, the WormLab parameters “Turn count” and “Wave initiation rate” were analyzed.
Generation of Tars1 mouse lines
The R432H variant was introduced into the mouse Tars1 locus using CRISPR-Cas9-mediated gene editing, which was performed by the University of Michigan Transgenic Animal Core. A single-stranded oligonucleotide (ssODN) was designed to introduce the R432H variant in cis with synonymous variants that ablated a BglI cut site that is present in the wild-type allele, and prevented binding of the guide RNA after repair. Cas9, sgRNA, and ssODN were injected into hybrid C57BL/6J × SJL/J F1 zygotes, which were implanted into pseudopregnant females. These mice produced 32 pups, which were genotyped by PCR amplification (primer sequences in Table S2) and BglI digestion to identify mice that had incorporated the repair template. Amplicons were submitted for Sanger sequencing to identify mice with proper integration of the repair template. These mice were mated to C57BL/6 mice to establish germline transmission. To assess the Mendelian ratios of offspring genotypes, 43 pups from crosses between Tars1^R432H/+^ females and Tars1^R432H/+^ males were genotyped using the BglI restriction enzyme digest strategy described above. Details for generating the F538Kfs∗4 allele can be found in the supplemental information.
Preparation of mouse tissues for histology
To investigate P0 pups for gross histological changes, dead pups were collected and live pups were killed by decapitation. Pups were individually fixed in neutral-buffered formalin, rocking overnight at room temperature. Pups were then placed in 70% ethanol and stored at 4°C. To investigate histological changes in the hair and skin of adult mice, three Tars1^R432H/F538Kfs∗4^ mice and their age-matched, sex-matched Tars1^R432H/+^ littermates were euthanized. The mice were shaved, and skin was collected from the dorsal trunk, ventral trunk, ears, tail, and paws, as well as from the area of the head with visible hair loss. Skin samples were placed on 0.45-μm HA filters (Millipore) wetted in PBS and strips were cut parallel to the direction of hair follicle growth. All strips were then fixed overnight in neutral-buffered formalin at room temperature, transferred to 70% ethanol, and stored at 4°C. Samples were shipped to Histoserv, Inc. for embedding and sectioning. Briefly, samples with bone were decalcified, tissues were dehydrated, and water inside of the tissues was replaced with paraffin wax. Tissues were then embedded into wax blocks of paraffin. Blocks were sectioned and affixed to slides (two sagittal sections were taken for the P0 pups). Adult skin sections were stained with H&E; P0 pup sections were stained with either H&E or PAS, which detects glycoproteins and mucins.
Analysis of epidermal thickness in P0 pups
Dorsal skin from H&E-stained sections was used to analyze the epidermal thickness of four Tars1^R432H/F538Kfs∗4^ mice and three Tars1^R432H/+^ littermates. Five 1-mm areas were selected, evenly spaced out across the back. In each 1-mm area, the thickness of the epidermis was measured by drawing lines in Adobe Illustrator that span the width of the epidermal layer, then using the 200-μm scale in each image to convert line length to microns. Five measurements were made that evenly spanned the 1-mm area; each measurement was made at the widest local area.
Data and code availability
The published article includes all datasets and code generated or analyzed during this study.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Antonellis A.Green E.D.The role of aminoacyl-t RNA synthetases in genetic diseases Hum. Genet.920088710710.1146/annurev.genom.9.081307.16420418767960 · doi ↗ · pubmed ↗
- 2Alexandrova J.Paulus C.Rudinger-Thirion J.Jossinet F.Frugier M.Elaborate u ORF/IRES features control expression and localization of human glycyl-t RNA synthetase RNA Biol.1220151301131310.1080/15476286.2015.108686626327585 PMC 4829322 · doi ↗ · pubmed ↗
- 3Tolkunova E.Park H.Xia J.King M.P.Davidson E.The Human Lysyl-t RNA Synthetase Gene Encodes Both the Cytoplasmic and Mitochondrial Enzymes by Means of an Unusual Alternative Splicing of the Primary Transcript J. Biol. Chem.2752000350633506910.1074/jbc.m 00626520010952987 · doi ↗ · pubmed ↗
- 4Meyer-Schuman R.Antonellis A.Emerging mechanisms of aminoacyl-t RNA synthetase mutations in recessive and dominant human disease Hum. Mol. Genet.262017 R 114R 12710.1093/hmg/ddx 23128633377 PMC 5886470 · doi ↗ · pubmed ↗
- 5Greco C.D.Antonellis A.The Role of Nuclear-Encoded Mitochondrial t RNA Charging Enzymes in Human Inherited Disease Genes 132022231910.3390/genes 1312231936553587 PMC 9777667 · doi ↗ · pubmed ↗
- 6Fuchs S.A.Schene I.F.Kok G.Jansen J.M.Nikkels P.G.J.van Gassen K.L.I.Terheggen-Lagro S.W.J.van der Crabben S.N.Hoeks S.E.Niers L.E.M.Aminoacyl-t RNA synthetase deficiencies in search of common themes Genet. Med.21201931933010.1038/s 41436-018-0048-y 29875423 PMC 7091658 · doi ↗ · pubmed ↗
- 7Dallabona C.Diodato D.Kevelam S.H.Haack T.B.Wong L.-J.Salomons G.S.Baruffini E.Melchionda L.Mariotti C.Strom T.M.Novel (ovario) leukodystrophy related to AARS 2 mutations Neurology 8220142063207110.1212/wnl.000000000000049724808023 PMC 4118500 · doi ↗ · pubmed ↗
- 8Steenweg M.E.Ghezzi D.Haack T.Abbink T.E.M.Martinelli D.van Berkel C.G.M.Bley A.Diogo L.Grillo E.NaudéJ.T.W.Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS 2 mutations Brain 13520121387139410.1093/brain/aws 07022492562 · doi ↗ · pubmed ↗