OxyR is required for oxidative stress resistance of the entomopathogenic bacterium Xenorhabdus nematophila and has a minor role during the bacterial interaction with its hosts
Victoria Bientz, Anne Lanois, Nadège Ginibre, Sylvie Pagès, Jean-Claude Ogier, Simon George, Stéphanie Rialle, Julien Brillard

TL;DR
This study shows that OxyR helps the bacterium Xenorhabdus nematophila resist oxidative stress and plays a minor role in its interaction with host nematodes and insect larvae.
Contribution
The study identifies the OxyR regulon in Xenorhabdus nematophila and its role in oxidative stress resistance and symbiotic interactions.
Findings
OxyR is crucial for in vitro oxidative stress resistance in Xenorhabdus nematophila.
An oxyR mutant showed no significant defect in reassociating with its nematode partner.
The oxyR mutant produced more offspring in the nemato-bacterial complex compared to the wild-type strain.
Abstract
Xenorhabdus nematophila is a Gram-negative bacterium, mutualistically associated with the soil nematode Steinernema carpocapsae, and this nemato-bacterial complex is parasitic for a broad spectrum of insects. The transcriptional regulator OxyR is widely conserved in bacteria and activates the transcription of a set of genes that influence cellular defence against oxidative stress. It is also involved in the virulence of several bacterial pathogens. The aim of this study was to identify the X. nematophila OxyR regulon and investigate its role in the bacterial life cycle. An oxyR mutant was constructed in X. nematophila and phenotypically characterized in vitro and in vivo after reassociation with its nematode partner. OxyR plays a major role during the X. nematophila resistance to oxidative stress in vitro. Transcriptome analysis allowed the identification of 59 genes differentially…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
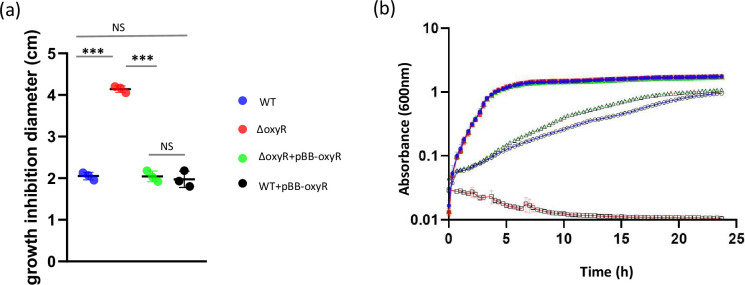 Fig. 1
Fig. 1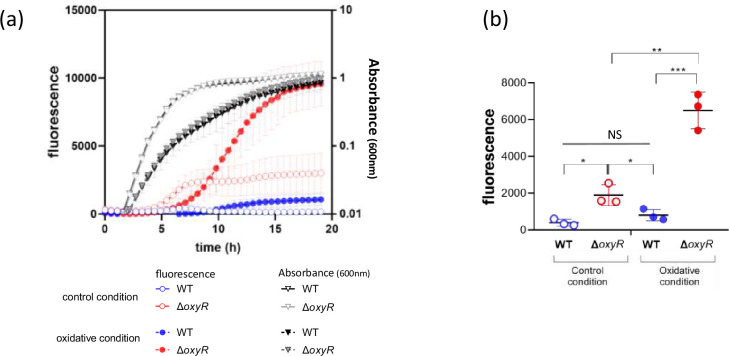 Fig. 2
Fig. 2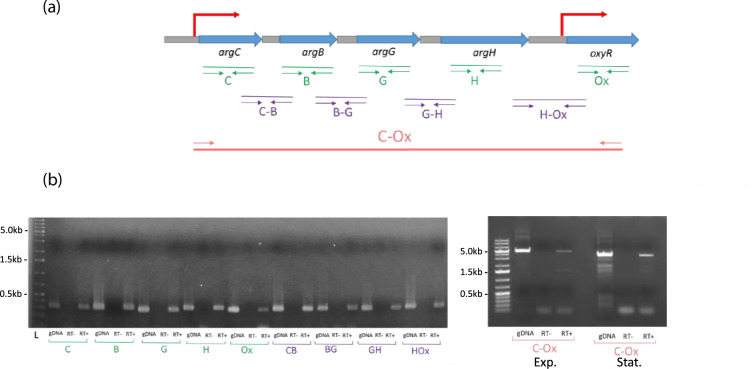 Fig. 3
Fig. 3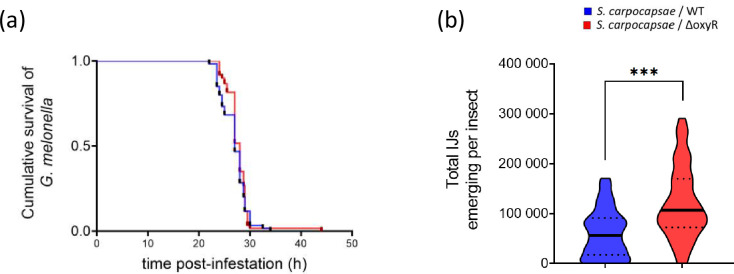 Fig. 4
Fig. 4| Strain or plasmid | Relevant genotype and characteristics* | Reference or source |
|
| ||
| Wild type | [ | |
|
| Agilent Technologies | |
|
|
| [ |
|
|
| This study |
|
| Wild type | Pasteur Institute Culture collection, Paris, France |
|
| ||
| pBBR1MCS-5 | Cloning vector, Gmr | [ |
| pBB-D3-gfp | pBBR1MCS-5 plasmid expressing the gfp-gene under the control of the strong and constitutively expressed D3 promoter | [ |
| pBB-oxyR | 1038 pb PCR fragment ( | This study |
| pHP45-ΩCm | Cmr cassette harbouring plasmid | [ |
| pJQ200KS | Mobilizable vector, Gmr | [ |
| pJQ-ΔoxyR | Region overlapping the | This study |
| pPROBE-gfp[AAV] | Plasmid harbouring a promoterless | [ |
| PoxyR’gfp[AAV] | pPROBE-gfp[AAV] expressing the GFP under the control of the | This study |
| Label | Gene name | Product | Mutant ΔoxyR/F1 wild-type strain | EGGNOG classification | |||||
| LB | LB +10 mM PQ | OG ID | Process | OG function | |||||
| log2 fold change | Adjusted | log2 fold change | Adjusted | ||||||
| XNC3_v3_0300 |
| Asparagine synthetase A | −0.32 | 3.78E-02 | −0.45 | 4.96E-02 | 05CU9 | Metabolism | Asparagine synthetase A |
| XNC3_v3_0367 | – | Conserved protein of unknown function | −0.31 | 3.78E-02 | – | – | 063XI | Information storage and processing | Toxic component of a toxin-antitoxin (TA) module. A |
| XNC3_v3_0507 |
| Acetylglutamate kinase | – | – | −0.42 | 2.89E-02 | 05CAS | Metabolism | nag kinase |
| XNC3_v3_0508 |
| Argininosuccinate synthase | – | – | −0.51 | 9.10E-03 | 05CDH | Metabolism | Citrulline--aspartate ligase |
| XNC3_v3_0509 |
| Argininosuccinate lyase | – | – | −0.73 | 4.83E-05 | 05CH7 | Metabolism | Arginosuccinase |
| XNC3_v3_0986 | – | Alkyl hydroperoxide reductase C | 1.54 | 4.41E-02 | – | – | 05D3R | Cellular processes and signalling | Alkyl hydroperoxide reductase |
| XNC3_v3_1095 | – | Conserved protein of unknown function | −0.55 | 4.96E-02 | – | – | 01ZBT | Metabolism | Ribosomal protein L11 methyltransferase (PrmA) |
| XNC3_v3_1410 | – | Conserved protein of unknown function | −1.01 | 1.42E-05 | −1.13 | 2.74E-03 | 07BCJ | Poorly characterized |
|
| XNC3_v3_1411 | – | DinI-like protein in retron EC67 | −0.85 | 3.43E-05 | – | – | 06GNW | Poorly characterized | DinI-like protein in retron EC67 |
| XNC3_v3_1412 | – | Tail protein X (GpX) | – | – | −1.19 | 5.44E-05 | 05YXG | Poorly characterized | Tail protein |
| XNC3_v3_1413 | – | Conserved protein of unknown function | −1.12 | 5.30E-03 | – | – | 060YZ | Poorly characterized |
|
| XNC3_v3_1414 | – | Conserved protein of unknown function | −0.81 | 1.48E-02 | −0.84 | 2.75E-03 | 05W5F | Poorly characterized | Bacteriophage Rz lysis protein |
| XNC3_v3_1415 | – | Baseplate assembly protein V (GpV) | – | – | −1.17 | 2.74E-05 | 05MCM | Poorly characterized | Baseplate assembly protein |
| XNC3_v3_1416 |
| Baseplate assembly protein W | – | – | −1.08 | 9.23E-04 | 05WAE | Poorly characterized | Baseplate assembly protein |
| XNC3_v3_1417 |
| Baseplate assembly protein J | −0.95 | 8.41E-05 | −1.11 | 1.34E-07 | 05F1E | Poorly characterized | Baseplate assembly protein |
| XNC3_v3_1418 | – | Tail protein I (GpI) | −1.00 | 2.52E-04 | −1.17 | 9.41E-06 | 06WW5 | Poorly characterized | Tail protein |
| XNC3_v3_1419 | – | Conserved protein of unknown function | −1.04 | 6.97E-06 | −1.11 | 2.51E-06 | 0638C | Poorly characterized | Tail fibre protein |
| XNC3_v3_1427 | – | Putative E14 prophage tail fibre protein (modular protein) | −0.76 | 2.66E-03 | −1.06 | 5.86E-06 | 06KCG | Poorly characterized | Phage tail collar domain |
| XNC3_v3_1428 |
| Major tail sheath protein XnpS1 of the xenorhabdicin, a R-type bacteriocin | – | – | −1.22 | 4.08E-06 | 07QJC | Poorly characterized | Tail sheath protein |
| XNC3_v3_1429 |
| Major tail tube protein XnpT1 of the xenorhabdicin, a R-type bacteriocin | – | – | −1.26 | 3.21E-06 | 08VRY | Poorly characterized | Tail tube protein |
| XNC3_v3_1430 | – | Conserved protein of unknown function | – | – | −1.17 | 5.65E-07 | 05W29 | Poorly characterized | Tail protein |
| XNC3_v3_1431 | – | Conserved protein of unknown function | −1.09 | 4.96E-02 | −1.35 | 8.42E-05 | 07FHP | Poorly characterized | TR |
| XNC3_v3_1432 | – | Conserved protein of unknown function | – | – | −1.26 | 4.12E-03 | 05E3X | Cellular processes and signalling | Lytic transglycosylase catalytic |
| XNC3_v3_1433 | – | Conserved protein of unknown function | −1.02 | 6.90E-07 | −1.06 | 8.62E-04 | 05NDM | Poorly characterized | P2 GpU family protein |
| XNC3_v3_1434 | – | Putative phage protein (D protein) (modular protein) | −0.91 | 4.80E-04 | −1.23 | 3.42E-05 | 05DAR | Poorly characterized | Late control |
| XNC3_v3_1435 |
| Prophage P2 transcriptional activator for bacteriophage P2 late genes | – | – | −1.01 | 8.62E-04 | 05ZTC | Information storage and processing | Transcriptional activator, Ogr delta |
| XNC3_v3_1715 | – | Putative alkylphosphonate uptake protein in phosphonate metabolism | – | – | −0.58 | 4.90E-03 | 08Z8G | Metabolism | Alkylphosphonate utilization operon protein PhnA |
| XNC3_v3_1716 |
| Fe-binding and storage protein | – | – | −3.24 | 2.86E-90 | 05DPV | Metabolism | Protects DNA from oxidative damage by sequestering intracellular Fe(2) ion and storing it in the form of Fe(3) |
| XNC3_v3_1779 |
| Thioredoxin reductase 1 | – | – | −0.37 | 7.43E-03 | 05C3M | Cellular processes and signalling | Thioredoxin reductase |
| XNC3_v3_2487 | – | Peptide synthetase | – | – | −0.79 | 8.08E-05 | 05C0W | Metabolism | Non-ribosomal peptide synthetase |
| XNC3_v3_2488 | – | Peptide synthetase | – | – | −0.90 | 5.71E-04 | COG1020 | Metabolism | Non-ribosomal peptide synthetase |
| XNC3_v3_2489 |
| Pyridine nucleotide transhydrogenase, beta subunit | – | – | −1.77 | 4.56E-13 | 05C15 | Metabolism | The transhydrogenation between NADH and NADP is coupled to respiration and ATP hydrolysis and functions as a proton pump across the membrane (by similarity) |
| XNC3_v3_2490 |
| Pyridine nucleotide transhydrogenase, alpha subunit | – | – | −1.54 | 2.63E-55 | 08IIE | Metabolism | The transhydrogenation between NADH and NADP is coupled to respiration and ATP hydrolysis and functions as a proton pump across the membrane (by similarity) |
| XNC3_v3_2825 | – | Conserved protein of unknown function | −2.17 | 8.56E-28 | −1.55 | 3.37E-19 | 08YW5 | Poorly characterized | Fimbrial protein |
| XNC3_v3_2826 |
| Putative outer membrane protein | −2.42 | 1.74E-33 | −1.49 | 5.21E-18 | 05CW0 | Cellular processes and signalling | Outer membrane usher protein |
| XNC3_v3_2827 |
| Putative fimbrial chaperone LpfB | −5.93 | 5.84E-77 | −5.42 | 4.92E-50 | 08VCD | Poorly characterized | Chaperone |
| XNC3_v3_2828 |
| Putative fimbrial subunit (pilin) | −6.41 | 3.80E-63 | −6.04 | 4.84E-114 | 05×0I | Cellular processes and signalling | Fimbrial |
| XNC3_v3_2903 | – | Protein of unknown function | −0.65 | 3.51E-03 | – | – |
| Unknown |
|
| XNC3_v3_3336 | – | Phage regulatory protein | −0.74 | 2.59E-02 | – | – | 06GH0 | Poorly characterized |
|
| XNC3_v3_3775 | – | Conserved protein of unknown function | −0.43 | 4.45E-03 | – | – |
| Unknown |
|
| XNC3_v3_3934 |
| Major MR/P fimbrial protein | – | – | −1.68 | 6.77E-08 | 05HMV | Cellular processes and signalling | Fimbrial protein |
| XNC3_v3_4172 |
| Glucosephosphate isomerase | – | – | −0.73 | 2.22E-05 | 07QP8 | Metabolism | Phosphohexose isomerase |
| XNC3_v3_4254 | – | Conserved protein of unknown function | −0.81 | 8.39E-03 | −0.97 | 1.15E-06 | 08KAS | Metabolism | Cholesterol oxidase |
| XNC3_v3_4306 | – | Conserved protein of unknown function | −1.14 | 8.39E-03 | −1.42 | 1.96E-03 | 07FAD | Poorly characterized |
|
| XNC3_v3_4313 | – | Putative major capsid protein | – | – | −1.61 | 3.33E-09 | 05E9P | Poorly characterized | Phage major capsid protein |
| XNC3_v3_4314 | – | Putative prohead protease | −1.52 | 5.69E-07 | −1.68 | 3.41E-12 | 05MAP | Poorly characterized | Phage prohead protease, HK97 family |
| XNC3_v3_4315 | – | Putative head portal protein | −1.30 | 2.35E-04 | −1.59 | 7.16E-11 | 05EFC | Cellular processes and signalling | Phage portal protein HK97 family |
| XNC3_v3_4316 | – | Putative head-tail adaptor | – | – | −1.54 | 2.70E-05 | 05×0P | Poorly characterized | Head-tail adaptor |
| XNC3_v3_4317 | – | Conserved protein of unknown function | – | – | −1.68 | 2.22E-05 | 069NN | Poorly characterized | Phage protein |
| XNC3_v3_4318 | – | HNH endonuclease | −1.47 | 4.80E-05 | −1.39 | 5.20E-05 | 05VK6 | Cellular processes and signalling | HNH endonuclease |
| XNC3_v3_4319 | – | Conserved protein of unknown function | – | – | −1.38 | 1.07E-03 | 0838W | Poorly characterized | Phage terminase small subunit |
| XNC3_v3_4320 | – | Putative terminase large subunit | – | – | −1.13 | 1.21E-02 | 05CAR | Information storage and processing | Terminase, large subunit |
| XNC3_v3_4482 | – | Putative head portal protein | −1.23 | 2.81E-03 | −1.04 | 6.92E-04 | 05EFC | Cellular processes and signalling | Phage portal protein HK97 family |
| XNC3_v3_4483 | – | Phage phi-C31 gp35-like protein | – | – | −1.16 | 1.10E-03 | 05MAP | Poorly characterized | Phage prohead protease, HK97 family |
| XNC3_v3_4484 | – | Phage major capsid protein, HK97 | −1.25 | 1.79E-03 | −1.33 | 3.05E-05 | 05E9P | Poorly characterized | Phage major capsid protein |
| XNC3_v3_4491 | – | Conserved protein of unknown function | – | – | −0.75 | 2.12E-02 |
| Unknown |
|
| XNC3_v3_4492 | – | Putative phage-related protein | – | – | −0.79 | 3.60E-02 | 067 WK | Information storage and processing | Transcriptional regulator |
| XNC3_v3_4545 | – | Conserved protein of unknown function | – | – | −0.56 | 1.21E-02 | 08Y1Q | Poorly characterized |
|
| XNC3_v3_4548 |
| Glutathione oxidoreductase | – | – | −2.00 | 1.01E-46 | 05DC8 | Metabolism | Reductase |
- —http://dx.doi.org/10.13039/501100008222Université de Montpellier
- —http://dx.doi.org/10.13039/501100001665Agence Nationale de la Recherche
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEntomopathogenic Microorganisms in Pest Control · Insect symbiosis and bacterial influences · Insect Resistance and Genetics
Data Availability
The datasets generated and analysed during the current study are available at Gene Expression Omnibus database using the accession number GSE244155.
Introduction
Bacteria have to cope with various stresses depending on the environmental conditions. Among those, bacterial redox stress can originate from exogenous factors, like abiotic environmental conditions or host response. Exogenous oxidative agents can enter the cells through the membranes by passive diffusion, and such influx can lead to an elevated cellular reactive oxygen species (ROS) level that causes an oxidative stress [1]. In addition, bacterial redox stress can originate from endogenously generated ROS since partially reduced forms of oxygen as well as hydroxyl radicals are formed continuously in aerobic bacterial cells [23]. ROS may lead to damage of intracellular macromolecules (DNA, RNA, proteins and lipids) in the cells. In order to avoid harmful effects of these oxidants, bacterial cells have to maintain them below a threshold of toxicity. The main strategy is to produce scavenging enzymes like catalases and peroxidases that degrade ROS. Because of putative damage to DNA, bacteria may also face higher mutation rates [4] and consequently elaborate a response involving activation of DNA repair enzymes [5]. Different redox-responsive transcriptional regulators can control the expression of numerous genes encoding enzymes involved in maintaining redox homeostasis [26]. Among them, the importance of OxyR in elaborating a response to mitigate ROS-mediated damages has been described in several bacteria [2].
Conserved in many Gram-negative and -positive bacteria [78], OxyR is a transcriptional regulator belonging to the LysR family that functions as a homotetramer [2]. It can act as an activator or as a repressor [79]. Depending on the redox state of the bacterial cell, the conformation of OxyR can change due to its disulfide bonds between two conserved cysteine residues, therefore allowing it to bind conserved motifs in the DNA [1011]. Additional modified forms of OxyR (S-nitrosylation, S-hydroxylation or S-glutathionylation) can occur in vivo and produce conformational changes in protein subunits, which yield differential DNA binding and transcriptional activity [12]. Moreover, the OxyR affinity for its DNA target site can also depend on the DNA methylation state, making this regulator one of the few described as involved in bacterial epigenetic regulation [1314]. Depending on the considered bacterial species, the size of the OxyR regulon can be broadly different, ranging from a few genes (four genes in Neisseria gonorrhoeae or five genes in Shewanella) to much more (122 genes identified in Pseudomonas aeruginosa) [1517]. In Escherichia coli, OxyR induces approximately two dozen genes, including katG (encoding a catalase), ahpCF [nicotinamide adenine dinucleotide (NADH) peroxidase], dps (DNA- and iron-binding protein), gorA (GSH reductase) and grxA (glutaredoxin), that allow to maintain the redox homeostasis [5]. Besides its function in bacterial adaptation to redox stress in vitro, OxyR has often been studied for its in vivo role in bacterial virulence, as described in several animal or plant pathogens [1819]. Studies involving oxyR knockout mutants have sometimes been tested in insect hosts: Yersinia pestis in Galleria mellonella [20], or P. aeruginosa in Drosophila melanogaster [21]. In these studies, while a role of OxyR in virulence has often been demonstrated, deletion of oxyR can also sometimes have no effect on bacterial-host interactions [22].
Xenorhabdus are entomopathogenic bacterial members of the Morganellaceae family. They are naturally found in a mutualistic symbiosis with soil nematodes belonging to the genus Steinernema. These nemato-bacterial complexes are able to infect and consequently kill many insect pests and can be used in biocontrol [23]. Xenorhabdus produce antimicrobial compounds in the insect host, which can prevent the multiplication of some soil or insect micro-organisms in the cadaver [24]. During their life cycle, Xenorhabdus have to switch between mutualism (within the nematode’s gut) and a pathogenic state (in the insect) [25]. After insect death, the bacteria, together with the nematodes, continue to multiply in the cadaver, during a necrotrophic stage [26]. The Xenorhabdus–Steinernema complexes are therefore useful model systems to study mechanisms of pathogenesis and mutualism [2527]. Gene deletion approaches have revealed the diversity of the mechanisms involved in the interactions between Xenorhabdus and its eukaryotic hosts. For instance, various factors were shown to contribute to pathogenesis (such as flagellar regulators [2829] or peptides [30]), to mutualism (e.g., the Mrx fimbriae [31]; Type 6 Secretion System components [32], lipase [33] and the nil genes [34]) or both (e.g., the CpxRA signal transduction system [35] and the global regulator Lrp [36]). Once inside the insect, in addition to its endogenously generated ROS, Xenorhabdus likely has to face H_2_O_2_ produced by the host, and presumably other ROS produced by various members of the microbiota [3738]. Up to now, the importance of oxidative stress encountered during Xenorhabdus lifecycle is not known. The role of OxyR in facing oxidative stress seems widely conserved in bacteria, but its diverse contribution to virulence depending on the bacterial pathogens, as well as the high variability in the OxyR regulon composition, has been reported. Here, we investigated the role of OxyR during the lifecycle of Xenorhabdus, a bacterial genus that lacks catalase activity [39]. Using a deletion mutant, the genes belonging to the OxyR-regulon were identified, and the OxyR role in X. nematophila was investigated in vitro during oxidative stress, as well as in vivo after reassociation of the bacterial strain with the Steinernema carpocapsae nematode.
Results
Deletion of the X. nematophila oxyR gene impairs growth in oxidative conditions
The XNC3_v3_0510 gene of X. nematophila strain F1 (annotated as oxyR) displays significant similarity with OxyR gene from several other bacterial species, including two cysteine residues (Cys199 and Cys208), described as involved in the formation of a disulfide bond under oxidizing conditions [4041]. A putative ribosome binding site sequence (CTGAGG) was found 8 Nt upstream of the ATG start codon. However, no clear OxyR binding site was predicted in the oxyR upstream region (www.prodoric.de). An oxyR deletion mutant was constructed and exposed to oxidative stress using paraquat (PQ). We first determined that PQ induces the formation of intracellular ROS in both the X. nematophila WT and the oxyR mutant strains. The ROS level was measured during growth in a standard Luria-Bertani (LB) medium, and no significant difference was observed between the WT and the OxyR mutant strains (Fig. S1, available in the online version of this article). As expected, addition of PQ in the growth medium increased the amount of ROS in the bacterial cells of both strains: 2.9-fold for the WT and 4.6-fold for the OxyR mutant. The X. nematophila growth in oxidative condition was then assessed on both solid and liquid media. Using a disc diffusion assay on agar medium, the halo of growth inhibition in the vicinity of the paper disc soaked with PQ (1M) was significantly larger for the oxyR mutant strain compared to that observed for the WT strain (P<0.05, t-test, Fig. 1a). In LB liquid medium (control condition), the growth curves of both strains had the same shape and overlapped: their slopes were similar during the exponential phase, and they reached the same maximum OD_600nm_ during stationary phase (Fig. 1b). When LB was supplemented with PQ (5 mM), a growth delay was observed for the WT strain, in comparison to LB control condition, revealing a growth impairment by PQ in X. nematophila. In the same oxidative condition, no growth was observed for the oxyR mutant. These results indicate that oxyR is required for X. nematophila growth in this oxidative stress condition. Complementation of the mutant strain was performed by introducing a low-copy number plasmid harbouring oxyR gene under a constitutively expressed promoter (pBB-oxyR*,* see Table 1). Results indicate that growth was restored by this complementation, to that of the X. nematophila WT strain, both on agar and in liquid media (Fig. 1a, b). Interestingly, the introduction of the same plasmid (pBB-oxyR) in the WT strain, which increased the number of oxyR copies in the bacterial cell and thus presumably increased the oxyR expression level, did not improve the X. nematophila resistance to this oxidative stress condition, since no reduction of halo size was observed on agar medium (Fig. 1a). This suggests that the overexpression of oxyR does not modify the X. nematophila resistance to oxidative stress.
The X. nematophila oxyR gene is involved in oxidative stress resistance. (a) Growth inhibition of X. nematophila F1 WT, ΔoxyR, ΔoxyR+pBB oxyR and WT+pBB oxyR by PQ (1M, 10 µL on a paper disc). Halo size of growth inhibition on LB agar was measured after 24 h. Data are the mean±SD of triplicate independent experiments (** significant difference, P<0.001; NS, no significant difference; t-test). (b) Growth of X. nematophila F1 WT (blue), ΔoxyR (red) and ΔoxyR+pBB oxyR (green) in oxidative stress condition (paraquat 5 mM, open symbols) or in control (LB, closed symbols). Absorbance at 600 nm was monitored for 24 h at 28 °C. Presented data are the mean values of three independent experiments, and error bars indicate standard deviations.*
Several other growth conditions were assessed to compare the phenotypes of the oxyR mutant to those of its parental strain. The growth curves of both strains were similar in LB (Fig. 1b). In addition, motility, biofilm formation ability and mutation rate were not significantly different between the two strains (Fig. S2). Other phenotypes usually performed to characterize Xenorhabdus strains were also investigated here on the oxyR mutant, and no significant difference was observed in comparison to the parental strain for bromothymol blue adsorption on NBTA (nutrient bromothymol blue adsorption medium), antibiotic production and lipase activities (Table S1). Altogether, these findings indicate that among the investigated phenotypes, only the susceptibility to PQ-induced oxidative stress is altered by the oxyR deletion in X. nematophila.
Characterization of the X. nematophila OxyR regulon
To study the expression of the OxyR regulon in X. nematophila, a transcriptomic approach was performed by RNA-seq analysis. WT and oxyR-mutant strains were grown in LB in exponential phase, and half of the culture was supplemented with PQ (10 mM) while the second one was used as a control. After 30 min of exposure to this oxidative agent, the cells were harvested for RNA sequencing and analysis. Data revealed a total of 59 genes that were significantly differentially expressed between the two strains (Table 2 and Fig. S3-A). All of them were downregulated in the mutant, except one upregulated in LB condition (XNC3_v3_0986 gene coding for an alkyl hydroperoxide reductase C). Twenty-one genes were differentially expressed at a significant level between the two strains regardless of the growth condition tested (i.e., in both LB control and oxidative condition), while 30 genes were differentially expressed between the two strains only in oxidative-stress condition. The genes belong to various functional groups according to their EGGNOG (Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups) classification, with many of them being phage-related genes, and the most represented category being ‘Metabolism’ (Table 2, Fig. S3-B). The expression level of some of these genes (argH, dps, trxB, pntA, yraJ, fimA and gor) was then validated using qRT-PCR (quantitative reverse transcription PCR) analysis. These seven genes were chosen because they were the most differentially expressed between the mutant and the WT strains, according to the RNA-seq data, except for argH which was slightly but significantly differentially expressed and was chosen because of its location in the vicinity of oxyR. Results confirmed the differential expression of each of these genes between the two strains (Fig. S4-A). A high correlation in the fold-change in expression was observed between the two techniques (RNA-seq and qRT-PCR) (Fig. S4-B).
The X. nematophila oxyR promoter is activated in the oxyR mutant
In order to investigate the activation of the presumed oxyR promoter, the intergenic region between the end of argH and the beginning of OxyR ORF (Fig. 3a) on the X. nematophila chromosome has cloned upstream the gfp[AAV] gene of pPROBE-gfp[AAV] vector (Table 1). This construct, pPoxyR’gfp[AAV], was transferred by mating into X. nematophila WT and the oxyR mutant. During growth of the strains harbouring the pPoxyR’gfp[AAV] transcriptional fusion, the observation of fluorescence confirmed the presence of an activated promoter located in this region (Fig. 2a). The level of fluorescence (mimicking the level of activation of the oxyR-promoter) was quantified during growth (Fig. 2a). In the LB-control condition, fluorescence was detected sooner and at a higher level in the oxyR mutant in comparison to that observed in the WT strain. During the exponential phase (at OD_600nm_ =0.5), the quantified fluorescence was about fourfold higher in the mutant than in the WT (1895 vs 402 units, Fig. 2b). When grown in oxidative conditions, the activation of the oxyR promoter was about eightfold higher in the mutant when compared to that of the WT (6504 vs 813 units, Fig. 2b). These results indicate that disruption of the chromosomal oxyR gene increased the activation of the plasmid-encoded oxyR promoter, revealing that the oxyR gene is negatively autoregulated in X. nematophila during these growth conditions. In addition, the activation of the oxyR promoter in the presence of PQ was higher for both strains compared to the standard (i.e., LB) condition (Fig. 2), indicating that growth in the oxidative condition activated the oxyR promoter.
*The promoter of the oxyR gene is downregulated by OxyR and is activated by PQ. Absorbance at 600 nm (shades of grey) and fluorescence (colours) of WT (blue) and ΔoxyR (red) strains carrying the PoxyRgfp[AAV] plasmid were quantified during growth in control (LB, open symbols) or oxidative stress condition (PQ = 5 mM, closed symbols). (a) Fluorescence measurement during growth of strains harbouring the PoxyRgfp[AAV] plasmid. (b) Fluorescence measurement when cells harbouring the PoxyRgfp[AAV] plasmid reached Abs600nm = 0.5. Error bars represent the standard deviation of the mean (n=3). Differences were significant at *, P<0.05; **, P<0.01; **, P<0.001 (t-tests). NS, no significant difference (P=0.1149).
The X. nematophila oxyR gene is co-transcribed with genes putatively involved in arginine metabolism. (a) Mapping of the primers used on the argC-oxyR locus. Thick blue arrows, ORF; grey boxes, intergenic regions. Thin green arrows, primers to amplify intragenic regions; thin purple arrows, primers to amplify intergenic regions. Thin red arrows, primers to amplify the whole locus, from argC to oxyR. Red broken arrows, putative promoters (schematic representation, not to scale). (b) RT-PCR on the argC-oxyR locus of X. nematophila. Left, primers were designed to amplify the intergenic regions between each gene of this locus (PCR product size ranged between 305 bp and 331 bp). RNA was extracted from X. nematophila harvested in exponential phase of growth. Right, primers were designed to amplify the entire locus from argC to oxyR (PCR product size 4915 bp). RNA was extracted from X. nematophila harvested in exponential phase (Exp.) and stationary phase (Stat.) of growth. For each primer couple, the wells correspond to the following: gDNA, positive control (PCR on genomic DNA); RT−, negative control (PCR on RNA without the reverse transcriptase step); RT+, RT-PCR on RNA with the reverse transcriptase step.
The X. nematophila oxyR gene is co-transcribed with upstream genes
RNA-seq analysis revealed that three of the four genes encoding proteins putatively involved in arginine metabolism (argCBGH), located upstream of oxyR in the same orientation (Fig. 3a), were significantly differentially expressed between the oxyR mutant and its parental strain, suggesting the existence of an operonic structure that may include the oxyR gene. In order to test this hypothesis, RT-PCR was performed on RNA extracted from exponential phase X. nematophila cells. For each of the five genes considered (argCBGH and oxyR), the detection of PCR-amplicons on RNA samples with a reverse transcriptase step indicated that they all were expressed in the tested condition. For each of these genes, the detection of mRNA overlapping its neighbouring gene was observed (Fig. 3b). In addition, using a primer pair mapping from argC to oxyR, a 4915 bp fragment was observed by RT-PCR amplification (while no amplification was observed in the negative control), indicating that the five genes were co-transcribed in the tested conditions (exponential and stationary phase of growth) and therefore constitute an operon (Fig. 3b). Considering the above-mentioned oxyR promoter detected by the observed fluorescence in t strains harbouring pPoxyR’gfp[AAV] (Fig. 2), all these results revealed that (at least) two independent promoters are able to activate oxyR transcription, one directly upstream from the oxyR gene (located between argH and oxyR) and one upstream from the argCBGH-oxyR operon (Fig. 3a).
Impact of the oxyR deletion on the bacterial lifecycle
The ability of the X. nematophila oxyR mutant to colonize its nematode host, Steinernema carpocapsae, an entomopathogenic nematode (EPN) was assessed. G. mellonella larvae were infected by axenized nematodes [infective juvenile (IJ) stage] and by either the X. nematophila oxyR mutant or the WT strain [both harbouring a plasmid allowing the constitutive expression of the GFP (Table 1)]. The bacterial and nematode partners were allowed to multiply in the insect cadavers, and the emerging IJs were collected after 30 days. Results presented in Fig. S5 revealed the presence of GFP-labelled bacteria in the receptacle of all the emerging IJs, indicating that both X. nematophila GFP-labelled strains (deleted of the oxyR gene or not) were able to colonize the nematodes. This qualitative approach was coupled to the quantification of the amount of bacterial cell per nematode using qPCR. Results revealed no significant difference in the calculated mean number of X. nematophila cells per IJ between the two nemato-bacterial complexes (Fig. S6). This indicates that deletion of oxyR does not impair the X. nematophila ability to re-associate to S. carpocapsae in a mutualistic relationship.
In order to determine if life history traits of the nemato-bacterial complexes might be modified by their reassociation with the bacterial strain mutated on the oxyR gene, we monitored the survival rate of G. mellonella larvae infested by 100 IJs over time. Results presented in Fig. 4a revealed that both nemato-bacterial complexes were pathogenic for the insects as they caused death in less than 48 h. No significant delay in killing G. mellonella larvae infested by the S. carpocapsae/X. nematophila ΔoxyR nemato-bacterial complex was observed throughout the cumulative survival curve, in comparison to that of the S. carpocapsae/X. nematophila WT nemato-bacterial complex (Fig. 4a). This suggests that OxyR is not required for full virulence in X. nematophila. The reproductive success of both these nemato-bacterial complexes was then assessed by quantifying the number of emerging nematodes from insect cadaver after 30 days post-infestation. The amount of IJs emerging from each G. mellonella cadaver was significantly higher with the S. carpocapsae/X. nematophila ΔoxyR nemato-bacterial complex compared to that of the S. carpocapsae/X. nematophila WT nemato-bacterial complex (Fig. 4b). This suggests that OxyR could play a role during the reproduction phase of the nematodes within the cadaver, affecting the number of offspring.
Impact of the X. nematophila oxyR deletion after bacterial reassociation with the S. carpocapsae nematode. Galleria mellonella larvae were infested by 100 S. carpocapsae IJs harbouring X. nematophila WT (blue) or oxyR mutant (red). Data represent three independent experiments, each containing 20 larvae. (a) Survival of G. mellonella larvae was monitored over time. The survival curves were not significantly different between the two strains (P=0.504 logrank test). (b) Emerging IJs from each G. mellonella cadaver 30 days after infestation. The amount of IJs exiting from each cadaver was significantly different between the two strains (t-test with Welch’s correction P<0.0001).
Discussion
The role of OxyR in X. nematophila, a bacterium mutualistically associated with an EPN, was investigated in the present study by constructing an oxyR mutant. While in some bacterial species, oxyR deletion can be associated with an impaired growth in control condition (i.e., without particular oxidative stress) compared to their parental strains, such as Bacteroides fragilis [42] and Haemophilus parasuis [43], and it most often does not affect the in vitro growth in others species, nor here in X. nematophila during growth in LB. In contrast, when exposed to oxidative conditions, the X. nematophila oxyR mutant exhibited a markedly elevated sensitivity compared to the parental WT strain. Such a role has also been described for numerous other bacterial species [2]. The generally conserved function of OxyR is to activate the expression of genes encoding for ROS-scavenging enzymes in response to oxidative agents. The sublethal concentrations of ROS that occur in the environment [44] can be mutagenic for bacteria, even in wild-type strains with functional oxidative stress responses [4546] indicating an inherent vulnerability in the bacterial ROS defence mechanisms [47]. In mutants that lack a functional oxyR gene, a higher mutagenesis rate can be expected, as reported in E. coli and Salmonella typhymurium [4850]. However, it is not always the case and may depend on the bacterial strain considered and/or on the tested growth conditions, since a frequency of spontaneous mutation similar between oxyR mutants and control strains has also been reported in other studies using the same species [5152]. The frequency of spontaneous mutations caused by endogenous ROS was investigated in X. nematophila after overnight growth in LB, and results revealed that oxyR deletion did not significantly cause a difference in the mutation rate, suggesting that no major oxidative stress is encountered and/or that internal mechanisms such as the global response to DNA damage (known as SOS response) are sufficiently efficient to repair DNA damages during this in vitro growth condition [5].
Among the other bacterial phenotypes associated with oxyR deletion, motility or biofilm formation has been reported in several species, as in Acidovorax citrulli [53] where OxyR positively regulates the expression of flagellin (FliC) and type IV pili (PilA) encoding genes. Here, the X. nematophila oxyR mutant exhibited no significant difference compared to the parental strain in its motility or biofilm-forming ability, as observed elsewhere [5455].
OxyR is a transcription factor that regulates several genes involved in anti-oxidative stress, such as catalase [2]. The description of the whole OxyR regulon was performed using an RNA-seq approach in X. nematophila in LB or in PQ-treated condition. Among the genes for which a differential level of expression was observed in the mutant compared to the parental strain, none display similarity with known catalase-encoding genes. This is in agreement with the fact that, in the Xenorhabdus genera, no catalase activity has been described [39], and no catalase-encoding gene has been identified [56]. However, some genes shown to belong to the OxyR regulon of other gamma-proteobacteria [57], such as dps, gorA, trxB and pntA, were also identified here. Some of them encode proteins with ROS-scavenging activities (NADH peroxidase; GSH reductase; a ferritin that lowers the availability of iron for Fenton reactions) [5859]. The gene ahpC, coding an alkylhydroperoxide reductase subunit C, is known to be activated by OxyR during oxidative conditions in many bacteria [759]. Surprisingly, our result suggests that expression of ahpC in X. nematophila is repressed by OxyR during non-oxidative stress condition, since this gene was the only one that was upregulated (1.5 fold) in the oxyR mutant compared to the WT in our conditions (during non-oxidative stress condition only). In the obligate anaerobic bacteria responsible for periodontitis, Tannerella forsythia [60] and Porphyromonas gingivalis [61], both being catalase negative similar as X. nematophila, ahpC is positively expressed by OxyR and this may compensate for the absence of catalase activity in peroxide detoxification. In Burkholderia thailandensis, ahpC was also shown to be negatively regulated by OxyR, while still helping to protect the bacteria from ROS [62]. Here, OxyR might indirectly regulate ahpC through the involvement of other regulatory proteins, leading to repression under non-oxidative conditions. Further studies are needed to elucidate the exact mechanisms underlying the ahpC regulation in X. nematophila.
Interestingly, a highly differentially expressed gene identified here during both oxidative-condition and control-condition, fimA encoding for a fimbria, was also found to belong to the OxyR regulon in P. gingivalis [61]. Fimbria are involved in the attachment and colonization of host tissues [63], and this finding may reveal a role of OxyR in the ability of X. nematophila to adhere to and colonize the nematode and/or insect’s tissues.
A high correlation in the fold-change expression was observed between the qRT-PCR and RNA-seq techniques. Altogether, these results enhance confidence in the list of the differentially expressed genes identified in this study, confirming the OxyR regulatory function in X. nematophila. Most differentially expressed genes were downregulated in the X. nematophila oxyR mutant, indicating a transcriptional activator role of OxyR in the tested conditions. Here, 59 genes were identified as differentially regulated in the X. nematophila oxyR mutant. This number is in the range of the OxyR regulon size described elsewhere, since it can vary significantly depending on bacterial species considered (from four genes in Neisseria gonorrhoeae [15] to hundreds of genes in H. parasuis [43]. The described sizes of OxyR regulon may also vary depending on the growth conditions in which the sampling for the analysis has been done, because the growth phase significantly contributes to bacterial resistance to a variety of stress conditions, including exposure to oxidative agents [6465].
Among the differentially expressed genes, those localized in the vicinity of oxyR and encoding putative enzymes involved in arginine metabolism were identified. RT-PCR experiments revealed that these genes were co-transcribed with oxyR and therefore belong to the same operon. A similar observation was made in E. coli [66]. The transcriptional fusion with the gfp reporter gene revealed a promoter located immediately upstream of oxyR. Our results revealed that the disruption of the chromosomal oxyR gene increased the activation of the plasmid-encoded oxyR promoter, suggesting that the oxyR gene is negatively autoregulated in X. nematophila. Whether this regulation is direct or not remains to be investigated, since this observation could potentially be explained by OxyR controlling another regulator, which then would activate the cloned promoter fragment of the pPoxyR’gfp[AAV] construct. Anyway, all these results indicate that at least two promoters can control oxyR transcription in X. nematophila. Interestingly, this synteny is conserved in the closely related genus Photorhabdus, but also in other gamma-proteobacteria such as E. coli [67]. Whether a similar co-transcription between oxyR and arginine metabolism-encoding genes also exists in these bacterial species and understanding the function of such co-transcription in the bacterial physiology remain to be investigated. Our RNA-seq data indicate that the argCBGH operon is overexpressed after addition of PQ when compared to control condition, for both the WT and the oxyR mutant strain. This reveals that the distal oxyR promoter (upstream of argC) is activated in oxidative condition in an OxyR-independent manner. Assessing whether differential regulation in oxidative conditions is similar or not in other bacterial species will require further studies.
Besides its in vitro involvement in bacterial adaptation to oxidative stress, several studies involving knockout mutants of bacterial pathogens revealed the contribution of OxyR in virulence. This was shown in mammalian pathogens, such as E. coli [19], Klebsiella pneumoniae [68], B. fragilis [42], Francisella tularensis [69] or P. aeruginosa [70] in various animal models, including insect hosts [2021]. OxyR can also contribute to virulence of plant pathogens such as Ralstonia solanacearum [18], Agrobacterium tumefaciens [71], Xanthomonas campestris [72] and Xanthomanas oryzae [73]. In addition, OxyR can also contribute in surviving the challenge of ROS encountered by some bacterial pathogens, during their free-living stages in the environment [58], such as Vibrio cholerae in seawater [74] or R. solanacearum in soil or water [75].
However, to our knowledge, there are no reports regarding the role of OxyR during a bacterial mutualistic association with its host. Because X. nematophila bacteria are specific symbionts of the nematode S. carpocapsae, we reassociated the oxyR mutant strain with its partner nematode. The pathogenicity of this successfully formed nemato-bacterial complex was then assessed in insect larvae, as during its natural life-cycle. The oxyR deletion in X. nematophila did not reveal a significant change in the ability of the nemato-bacterial complex to kill G. mellonella larvae. Deletion of oxyR has also been shown in other bacterial species to have no effect in virulence, as in Corynebacterium diphtheriae [76], Neisseria gonorrhoeae [15] or Mycobacterium maritimus [22].
Because we have shown that X. nematophila oxyR mutant displayed impaired growth during in vitro oxidative conditions, these in vivo results therefore suggest that additional factors, other than the X. nematophila OxyR, may help the nemato-bacterial complex to cope with oxidative stress encountered during the interaction of these multiple organisms (insect-nematode-microbiota). Indeed, oxidative stress can be an important immune mechanism used by insect to resist microbial infections (as shown in * G. mellonella* after infection by Salmonella enterica [37]). A possible explanation is the efficiency of the nemato-bacterial complex to avoid the effects of the insect host immune system: X. nematophila produces several cytolysines that may prevent phagocytosis and nodulation processes [77], as well as metabolites able to inhibit the phenoloxidase cascade [78], and the cognate nematode is also able to compromise host immunity [79]. The role played by the whole bacterial microbiota should also be considered [80]. An additional explanation could be alternative regulators that may allow to elicit an anti-oxidative response in X. nematophila during the infectious process. In bacteria, the defence system used to counter oxidative stress can be orchestrated by several transcriptional factors with overlapping functions, including OxyR [8182]. The importance of such additional regulators (SoxS is found in the X. nematophila genome for instance) when coping with oxidative stress in vivo remains to be explored. In addition, we cannot rule out the existence of an host compound that would directly interact with the OxyR protein, as described for the plant pathogen Xanthomonas [83], therefore leading to no phenotypic differences between the WT and the oxyR mutant strains.
In our study, this tripartite experiment also allowed us to investigate the impact of oxyR deletion during X. nematophila mutualistic interaction with the nematode. As mentioned above, both the WT and the mutant bacterial strains were able to reassociate with S. carpocapsae, with a similar level of colonization, revealing that they succeeded in supporting the nematode development. Interestingly, since a slightly increased reproductive success (measured as the number of offspring emerging from the insect cadaver) was observed for the nemato-bacterial complex harbouring the X. nematophila oxyR mutant, our results suggest that OxyR in X. nematophila may play a role in the nematode’s fitness in the G. mellonella model. However, we cannot rule out that this phenotype is linked to OxyR in an indirect manner. An additional experiment using axenized EPNs reassociated to a ΔoxyR complemented strain would be required to confirm if the WT phenotype is restored in vivo. Given that trophic analyses demonstrate that the nematode feeds on the bacteria and that the bacteria are responsible for consumption of the insect [84], this may suggest a slightly higher availability of the nutrients for the nematode when feeding on the X. nematophila oxyR mutant. Interestingly, a tween-specific lipase was also shown to play a role in supporting production of nematode progeny in G. mellonella [33], emphasizing the fact that nutrient availability in insect cadaver is important for EPN reproductive success. The observation of several bacteriocin or phage-related genes differentially regulated in the oxyR mutant compared to the WT in our RNA-seq data has been observed elsewhere [8587]. Considering the EPN as holobionts, future studies may also consider oxidative stress, since changes in OxyR regulation of phage-related genes could impact the associated microbiota composition and possibly the EPN life-cycle [88]. Studying all these interactions may provide new insights to better understand their efficiency in killing insect pests and therefore enhance their use in biocontrol.
In conclusion, we report here that oxyR is required for X. nematophila optimal growth in in vitro ROS-enriched environments but not during interactions with its eukaryotic hosts. This bacterial species lacks a catalase-encoding gene, but several genes that presumably contribute to the oxidative stress response could be identified as members of the OxyR regulon. Altogether, our results suggest that factors other than OxyR could allow X. nematophila to cope with oxidative stress encountered during the insect infection. Future studies should consider the importance of OxyR during bacterial mutualistic interactions with their host.
Methods
Strains and growth conditions
The strains used in this study are listed in Table 1. The X. nematophila F1 and E. coli bacterial strains were routinely grown in LB medium with a 180 r.p.m. agitation at 28 °C or 37 °C, respectively. As required, antibiotic concentrations used for bacterial selection were chloramphenicol 15 μg mL^−1^, gentamycin at 15 μg mL^−1^, rifampicin at 50 μg mL^−1^ and kanamycin at 20 μg mL^−1^. The oxidative agent PQ (also known as methyl viologen dichloride hydrate, Sigma-Aldrich) was added when required, at concentrations to either abolish the growth or to allow a remaining (impaired) growth of the mutant strain, and was based on similar studies in other bacterial species [15].
Bromothymol blue adsorption; antibiotic production against Micrococcus luteus; lipase activity on Tween 20, 40, 60, 80 and 85; and hemolytic activity were determined as previously described [89; 90]. For motility assays, agar plates were prepared with LB broth supplemented with 0.35 % agar and inoculated using 5 µL of cells grown in exponential phase (OD_600nm_ = 0.8), as previously described [91]. The diameter of the halo size of swimming motility was measured 20 h after incubation. Data from three independent experiments (with 10 plates used in each condition) were analysed using the Wilcoxon test. The spontaneous mutation rate was assessed by quantifying the emergence of rifampicin-resistant c.f.u.s, as previously described [92]. Briefly, strains were grown overnight in 40 mL of LB medium to reach a population above 1×10^9^ c.f.u. mL^−1^. Bacterial cells were serially diluted and plated on GNO +/-rifampicin (50 µg mL^−1^). The mutation rate was calculated as the rifampicin-resistant population divided by the total population. Data from three independent experiments were compared and analysed by t-test. Biofilm formation was monitored as previously described [92]. Briefly, 5 mL of LB medium in glass tubes was inoculated with an overnight culture and incubated for 12 days at 28 °C in static conditions. The tubes were then rinsed with PBS before the addition of 7 mL of crystal violet solution at 0.01% (in PBS) to stain the biofilms for 15 min. Biofilms were rinsed with PBS and then dissolved for 3 h in 7 mL of ethanol. The OD_570 nm_ measurement allowed the quantification of the biofilm-associated crystal violet. Data from three independent experiments with replicates (totalizing 30 tubes for each strain) were analysed using a Wilcoxon test.
Resistance to oxidative stress was quantified using PQ (paraquat dichloride hydrate, Sigma), as follows. A 6 mm paper disc (Millipore) soaked with 10 µL of PQ (1M) was placed on LB agar medium previously inoculated with 1 mL of exponential phase-grown bacterial cells (OD_600nm_ = 0.6). Halo size of growth inhibition was measured after 24 h. For each of the three independent experiments, technical triplicates were performed. Presented data were analysed using a Student’s t-test. In liquid medium, growth of X. nematophila strains in LB, or LB supplemented to 5 mM PQ, was monitored with a TECAN automated turbidimetric system (Infinite M200 TECAN), as previously described [92]. Experiments were performed on three independent biological replicates for each strain.
Cellular ROS level was quantified by fluorescence spectroscopy as previously described [93]. Briefly, X. nematophila cells were grown in exponential phase. At OD_600nm_ = 0.3, PQ (2.5 mM) was added (or not for the control condition), and growth was resumed for 30 min. Cells were then harvested by centrifugation (2 min at 13 000 r.p.m.) and washed twice in PBS buffer, followed by an incubation step of 30 min with DCFH-DA (2,7-dichlorofluorescein-diacetate at 100 µM, D6883, Sigma-Aldrich). Fluorescence was measured using a microplate reader (excitation wavelength 485 nm, emission wavelength 535 nm). Each independent experiment was performed in technical triplicate. The presented data are from three independent experiments and were analysed using t-test.
X. nematophila wild-type and ∆oxyR strains carrying pPoxyR’gfp[AAV] (Table 1) were cultured in black-sided, clear-bottomed 96-well plates (Greiner) as follows. For each well, 10 µL of an overnight culture diluted 1/40 was added to 190 µL of LB supplemented with kanamycin, with or without PQ as required. The plates were incubated in an Infinite M200 microplate reader (Tecan) at 28 °C with shaking. The OD_600nm_ and the GFP fluorescence (excitation 485 nm, emission 535 nm) were monitored every 30 min during 20 h. Three biological independent replicates were carried out for each condition.
Cloning and molecular biology
Chromosomal DNA was extracted from bacterial cells using the QIAamp DNA Mini kit (Qiagen, Courtaboeuf, France). The extraction of plasmid DNA from E. coli was performed using the GenElute™HP Plasmid miniprep purification kit as recommended by the manufacturer (Sigma, Saint-Quentin-Fallavier, France). Restriction enzymes and T4 DNA ligase were used as recommended by the manufacturers (New England Biolabs, Evry, France and Promega, La Farlede, France, respectively). Oligonucleotide primers sequences were designed using the Primer3 software [94]. They were synthesized by Integrated DNA Technologies (IDT, Leuven, Belgium) and are listed in Table S2. PCR was performed in a T100 thermal cycler (Biorad, Marnes-la-Coquette, France) using the iProof high-fidelity DNA polymerase (Biorad). Amplified DNA fragments were purified using a PCR purification kit (Ozyme, St Cyr LEcole, France) and separated on 0.7 % agarose gels after digestion as previously described [95]. Digested DNA fragments were extracted from agarose gels with a centrifugal filter device (Ozyme). All constructions were confirmed by DNA sequencing (Eurofins Genomics, Ebersberg, Germany).
Construction of the oxyR mutant was performed as follows. DNA fragments of the oxyR (XNC3_v3_0510) upstream (650 pb) and downstream (630 bp) regions were PCR-amplified using the primer pairs upF-oxyR-Pst/upR-oxyR-Sal and dnF-oxyR-Sac/dnR-oxyR-Xba, respectively (Table S2). PCR products were digested with PstI/SalI and SacI/XbaI using the primer-incorporated restriction sites. In parallel, a Cm^r^ cassette was PCR amplified from pHP45-ΩCm (Table 1), using the primer pairs Cm-F/Cm-R (Table S2) and digested with SalI/SacI, as previously described [96]. The three digested DNA fragments were purified, ligated in PstI/XbaI-digested pJQ200KS (Table 1) and introduced by electroporation in E. coli XL1. The resulting pJQ-ΔoxyR plasmid was transferred in the E. coli donor strain WM3064 and then introduced in X. nematophila by conjugative mating as previously described [28]. Four independent transconjugant clones were isolated and subjected to allelic exchange in LB at 28 °C, following the protocol routinely used in the laboratory [9698]. Clones of oxyR mutants were positively selected on sucrose plate and verified by PCR using VerifDoxyR-F and VerifDoxyR-R primers (Table S2) and sequencing. Furthermore, no polar effect of the deletion was observed according to the RNA-seq data showing no difference in expression of the oxyR surrounding genes in LB-control condition, between the WT and the mutant strain (Table 2).
Cloning the X. nematophila oxyR gene under Plac promoter was performed as follows. A 1038 bp DNA fragment was PCR amplified using two primers mapping upstream and downstream (Cp-oxyR -F/Cp-oxyR-R, respectively, Table S2) of the oxyR gene, using the following cycling conditions: 95 °C, 10 s; 56 °C, 30 s; 72 °C, 45 s for 35 cycles. It was then digested according to the endonuclease sites introduced in the primers (EcoRI/BamHI) and was inserted between the corresponding sites of the low-copy plasmid pBBR1MCS-5 (Table 1) downstream of the Plac promoter. The recombinant plasmid (pBB-oxyR) was introduced in E. coli WM3064 donor strain and then transferred in X. nematophila as described above. Transconjugants harbouring the pBBR1MCS-5 empty plasmid were used as a control. The construction of a transcriptional fusion between the upstream oxyR region and the gfp-reporter gene was performed as follows: DNA fragment harbouring putative promoter was amplified by PCR from X. nematophila F1 genomic DNA using the primer pairs PoxyR-F-Xba/PoxyR-R-Eco (Table S2). The PCR products were purified and digested with EcoRI and XbaI and inserted into the corresponding sites of pPROBE-*gfp[*AAV], a plasmid encoding a promoterless gfp-reporter gene (Table 1). The plasmids were transferred to X. nematophila by conjugative mating as described above.
RNA extractions, sequencing and amplification assays
X. nematophila wild-type and ∆oxyR strains were grown in 20 mL LB at 28 °C with shaking, after inoculation by an overnight growth culture at 0.5 %. When OD_600nm_ reached 0.3, the culture was split into two parts, and one of them was supplemented with PQ (10 mM) after which growth was immediately resumed for 30 min to generate an oxidative stress. The second one was used as a control. Four millilitres were then mixed with 8 mL of RNA-protect solution according to the manufacturer’s instructions (Qiagen). Total RNA extraction was performed using RNeasy miniprep Kit (Qiagen), with an additional incubation step with TURBO DNase (Invitrogen) as previously described [99]. The quantity and quality of RNA were assessed with an Agilent 2100 Bioanalyzer with the RNA 6000 Nano LabChip kit. The lack of DNA contamination was controlled by carrying out a PCR using 63bis and 153Rev primers mapping the 16S region (Table S2) on each RNA preparation. Nine independent cultures with cells harvested in exponential phase were performed for each strain and each growth condition.
For the RNA sequencing, equal amounts of total RNA from three independent samples per strain and per condition were pooled together to generate one final biological RNA sample per strain. Thus, from the initial nine independent RNA samples per strain and per condition, three final RNA samples were generated for each strain and subsequently treated as follows prior sequencing. MicrobExpress Bacterial mRNA Enrichment Kit (Ambion) was used to remove ribosomal RNA from 4 µg of total RNA according to the manufacturer’s instructions.
RNA-seq libraries were prepared using the Truseq stranded mRNA sample prep kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Briefly, the RNAs, previously depleted in rRNA, were fragmented using divalent cations at elevated temperature and reverse transcribed using random hexamers, SuperScript II (Thermo Fisher Scientific, Carlsbad, CA) and actinomycin D. Deoxy-TTP was replaced by dUTP during the second strand synthesis to prevent its amplification by PCR. Double stranded cDNAs were adenylated at their 3' ends and ligated to Illumina’s adapters containing unique dual indexes (UDIs). Ligated cDNAs were PCR amplified for 15 cycles PCR and the PCR products were purified using AMPure XP Beads (Beckman Coulter Genomics, Brea, CA, USA). The size distribution of the resulting libraries was monitored using a Fragment Analyzer (Agilent Technologies, Santa Clara, CA, USA), and the libraries were quantified using the KAPA Library quantification kit (Roche, Basel, Switzerland). The libraries were denatured with NaOH, neutralized with Tris-HCl and diluted to 300 pM. Clustering and sequencing were performed on a NovaSeq 6000 (Illumina, San Diego, CA, USA) using the single-end 100 nt protocol on one lane of a flow cell SP.
Image analyses and base calling were performed using the NovaSeq Control Software and the Real-Time Analysis component (Illumina, San Diego, CA, USA). Demultiplexing was performed using Illumina’s conversion software (bcl2fastq 2.20). The quality of the raw data was assessed using FastQC (v0.11.8) from the Babraham Institute and the Illumina software SAV (Sequencing Analysis Viewer). FastqScreen (v0.14.0) was used to identify potential contamination.
Reads were aligned to the reference genome (X. nematophila XENF1.2) with BWA-MEM/BWA-ALN (v0.7.17-r1188) [100]. Reads with MAPQ scores less than three were filtered out using SAMtools view (v1.9, options -F 0×904 q 3) to eliminate the reads that were likely to align to multiple positions. SAMtools was also used to sort and index the alignment files. Then, gene counting was performed with featureCounts v2.0.0 [101] using a gff file downloaded on 27 July 2020 from the MaGe platform (https://mage.genoscope.cns.fr) [102]. As the data are from a strand-specific assay, the reads have to be mapped to the opposite strand of the gene (-s two option). Before statistical analysis, genes with less than 15 reads (cumulating all the analysed samples) were filtered out.
Differentially expressed genes were identified using the Bioconductor [103] package DESeq2 v1.26.0 [104] (R version 3.6.2). Data were normalized using the DESeq2 normalization method. Genes with adjusted p-value below 5 % (according to the FDR method from Benjamini-Hochberg) were called differentially expressed.
The complete dataset from this study has been deposited in NCBI’s Gene Expression Omnibus database, under accession number GSE244155.
RT-PCR
RNA from X. nematophila cells grown in exponential phase (OD_600nm_ =0.4) or stationary phase (OD_600nm_=2.5) was extracted as described above. Lack of DNA contamination was controlled by carrying out a PCR on 16S gene using 63bis and 153Rev primers (Table S2) on each RNA preparation as previously described [90]. RT-PCR reactions were performed as follows. A total of 500 ng of total RNA was denatured at 70 °C for 5 min. Conversion into cDNA was carried out using random hexamers (C1181, Promega) and SuperScript II reverse transcriptase (Invitrogen), for 10 min at 25 °C, then 50 min at 42 °C and finally 15 min at 70 °C. PCR amplifications on the cDNA were carried out with a Go-Taq DNA polymerase (Promega) using primers listed in Table S2. The following amplification conditions were applied: 3 min at 95 °C, 30 s at 55 °C and 15 min at 72 °C, for 35 cycles. Positive controls were performed using X. nematophila F1 genomic DNA as template. RNA samples that have not undergone the reverse transcriptase treatment were used as negative controls. The PCR products were loaded on an agarose gel followed by visualization on a UV transilluminator.
RT-qPCR analysis
Quantitative reverse transcription-PCR (RT-qPCR) was carried out as previously described [90] on seven genes selected because they were highly differentially expressed between the WT and oxyR mutant strains according to the RNA-seq analysis. Briefly, RNA samples from three biological replicates for each strain and each growth condition were used for cDNA synthesis as described above. The SuperScript II reverse transcriptase (Invitrogen) was used on 0.5 µg of total RNA with random hexamers (100 ng·µL^−1^; Roche Diagnostics). qPCR analyses were performed using SensiFAST™ SYBR No-ROX Kit (Bioline) with 0.5 µL of cDNA synthesis mixture (diluted 1:50) and specific gene primers at 1 µM (Table S2). The reactions were performed in duplicate at 95 °C for 2 min, followed by 45 cycles at 95 °C for 5 s, 61 °C for 25 s and 70 °C for 15 s and monitored in the LightCycler 480 system (Roche). Melting curves were analysed and always contained a single peak. The data were analysed with the REST software 2009 [105] using the pair-wise fixed randomization test with 5000 permutations. Data are presented as a ratio with respect to the reference housekeeping gene recA, as previously described [90].
Bacterial reassociation with axenic nematodes
S.carpocapsae (strain SK27) axenic nematodes were produced as previously described [106]. Briefly, 4–6 days after insect infestation with L3 (infective juvenile, IJ), gravid female nematodes were collected from G. mellonella cadaver. Eggs were extracted from washed female by crushing. The intact eggs (checked by microscopic observation) were placed on sterile liver-agar supplemented with antibiotics. After 30 days in a sterile, dark and humid environment, axenic IJs were collected. The absence of Xenorhabdus in the axenized nematodes was verified by DNA extraction followed by PCR amplification with Xenorhabdus-specific primers (Xeno_F/Xeno_R, Table S2) that target a region of the XNC1_0073 gene encoding a putative TonB-dependent haem receptor [107].
X. nematophila WT and ∆oxyR strains harbouring a plasmid expressing GFP under the control of a constitutive promoter (pBB-D3-gfp, Table 1) were obtained using the mating protocol described above. They were grown in LB until an OD_600nm_ of 0.6, harvested and washed in fresh LB. Approximately 10**^7^** c.f.u. in 20 µL LB supplemented with kanamycin were injected into G. mellonella larvae (five larvae for each strain) in order to initiate the in vivo bacterial growth. After 6 h, insects were still alive and were infested by axenic IJs, in order to allow a multiplication of the nematodes inside the infected insect larvae. The infestations were performed by pipetting 100 axenic IJs onto each immobilized G. mellonella larvae in 1.5 mL Eppendorf tubes, as previously described [108]. The new generation of IJs that emerged was collected, and the presence of X. nematophila WT or ΔoxyR expressing the GFP in the IJ’s receptacle was confirmed under a fluorescence microscope (Fig. S5). The nematodes were stored in the dark at 9 °C.
Nemato-bacterial infestation
Twenty G. mellonella larvae were individually infested by 100 IJs and were then incubated in the dark at 23 °C. Three independent replicates were performed (totalizing 60 insect larvae for each nemato-bacterial complex). Insect survival was monitored over time to evaluate the virulence-related properties of each nemato-bacterial complex up to 45 h after infestation. Data were analysed in GraphPad Prism 8 software using the log-rank method with 95 % CI. Dead larvae were then placed individually on white traps [109]. After up to 30 days, the emergence of nematodes was observed for all dead larvae. To evaluate the fitness of the nematodes, the reproductive success (defined as the number of IJs that emerged from each insect larvae) was quantified after 30 days of infestation. As the reproductive success of nematodes is neither normally distributed nor normalizable, data were analysed using a Mann–Whitney test as previously described [108].
Quantification of Xenorhabdus smbiotically associated with S. carpocapsae
DNA was extracted from S. carpocapsae SK27 ApoIII/WT and ApoIII/∆oxyR (IJs) as previously described [80], with the following modifications. First, 100 IJs harbouring the Gfp-labelled X. nematophila strains were individually collected with a sterile pipette, transferred into screw-top microtubes (2 mL) and immediately frozen at − 80 °C. A heat stress (20 min incubation at +80 °C after their removal from the freezer) allowed to weaken the IJs’ double cuticle. Then, six 2-mm glass beads were added in each tube, and nematodes were mechanically disrupted using the FastPrep instrument (MP Biomedicals, Illkirch-Graffenstaden, France) for 3 cycles of 7 m/s of 40 s. One hundred microlitres of Quick Extract lysis solution (Bacterial DNA Extraction Kit from Epi-centre, USA), 1 µL of Ready-Lyse Lysozyme Solution (Epi-centre, USA) and 20 µL EDTA (0.5 M, pH 8) were added, and complete lysis of both nematodes and prokaryotic cells was obtained by incubation at room temperature for 90 min. Ten microlitres of Proteinase K (20 mg mL^−1^) was added followed by incubation at 55 °C with shaking until the solution was cleared. RNA was eliminated with 10 µL of RNaseA at 20 mg mL^−1^ (Invitrogen PureLink™ RNaseA, France) for 15 min at 37 °C. Proteins were precipitated by adding 100 µL of protein precipitation solution (Kit Wizard, Promega, France) followed by an incubation time of at least 5 min on ice; the samples were then centrifuged 5 min at 17 000 g at 4 °C, and the supernatant was collected. DNA was precipitated with isopropanol, resuspended in 50 µL ultrapure water and stored at −20 °C. The amount of Xenorhabdus in each IJ was then quantified using Xenorhabdus-specific primers (Xeno_F/Xeno_R, Table S2) as previously described [107]. Axenic nematode without Xenorhabdus were used as a control. Data from three independent samples were compared using the t-test with Welch’s correction.
supplementary material
10.1099/mic.0.001481Uncited Supplementary Material 1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Imlay JA Cellular defenses against superoxide and hydrogen peroxide Annu Rev Biochem 20087775577610.1146/annurev.biochem.77.061606.16105518173371 PMC 3057177 · doi ↗ · pubmed ↗
- 2Imlay JA Transcription factors that defend bacteria against reactive oxygen species Annu Rev Microbiol 2015699310810.1146/annurev-micro-091014-10432226070785 PMC 4618077 · doi ↗ · pubmed ↗
- 3Korshunov S Imlay JA Two sources of endogenous hydrogen peroxide in Escherichia coli Mol Microbiol 2010751389140110.1111/j.1365-2958.2010.07059.x 20149100 PMC 3049997 · doi ↗ · pubmed ↗
- 4Imlay JA Linn S Mutagenesis and stress responses induced in Escherichia coli by hydrogen peroxide J Bacteriol 19871692967297610.1128/jb.169.7.2967-2976.19873298208 PMC 212335 · doi ↗ · pubmed ↗
- 5Gupta A Imlay JA Escherichia coli induces DNA repair enzymes to protect itself from low-grade hydrogen peroxide stress Mol Microbiol 202211775476910.1111/mmi.1487034942039 PMC 9018492 · doi ↗ · pubmed ↗
- 6Sevilla E Bes MT González A Peleato ML Fillat MF Redox-based transcriptional regulation in prokaryotes: revisiting model mechanisms Antioxid Redox Signal 2019301651169610.1089/ars.2017.744230073850 · doi ↗ · pubmed ↗
- 7Chiang SM Schellhorn HE Regulators of oxidative stress response genes in Escherichia coli and their functional conservation in bacteria Arch Biochem Biophys 201252516116910.1016/j.abb.2012.02.00722381957 · doi ↗ · pubmed ↗
- 8Méndez V Rodríguez-Castro L Durán RE Padrón G Seeger M The Oxy R and Sox R transcriptional regulators are involved in a broad oxidative stress response in Paraburkholderia xenovorans LB 400Biol Res 202255710.1186/s 40659-022-00373-735184754 PMC 8859910 · doi ↗ · pubmed ↗