Construction of the Bicyclic Carbon Framework of Euphosalicin
David Schachamayr, Johanna Templ, Matthias Weil, Peter Gaertner, Valentin S. Enev

TL;DR
This paper describes the synthesis of a complex natural compound called euphosalicin, focusing on constructing its carbon framework.
Contribution
The paper presents a novel synthetic route to the C11/C12 (Z) isomer of the euphosalicin skeleton.
Findings
Asymmetric dihydroxylations and ring-closing enyne metatheses were successfully applied in the synthesis.
Highly advanced precursors for macrocyclization studies were isolated.
The unique C11/C12 (Z) isomer of the euphosalicin skeleton was prepared.
Abstract
Our studies toward the total synthesis of the natural product euphosalicin (1) are presented. Different approaches targeting key intermediates are described, the synthesis of which includes findings on asymmetric dihydroxylations and ring-closing enyne metatheses (RCEYM). Their connection allowed the isolation of highly advanced precursors for studies on macrocyclizations. Our efforts culminated in the preparation of the unique C11/C12 (Z) isomer of the C13 nor methyl skeleton of euphosalicin (1).
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Scheme 1
Scheme 1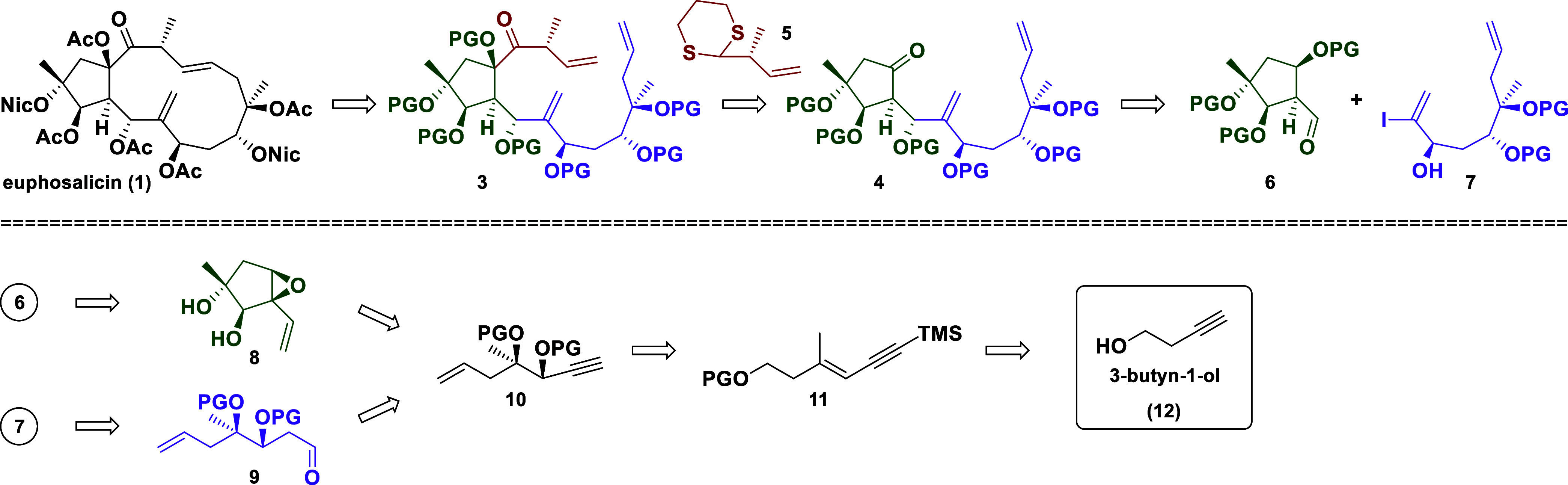 Scheme 2
Scheme 2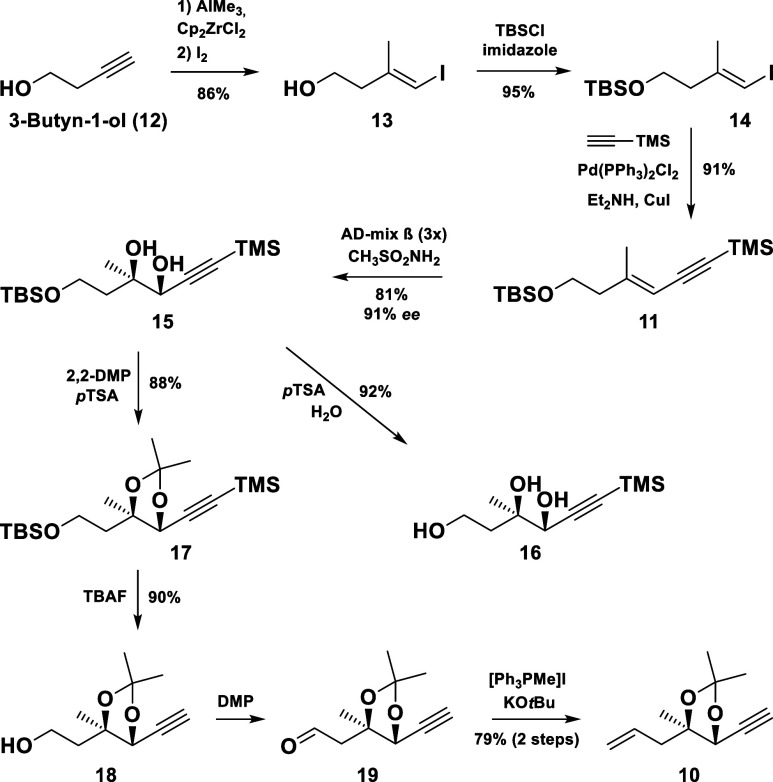 Scheme 3
Scheme 3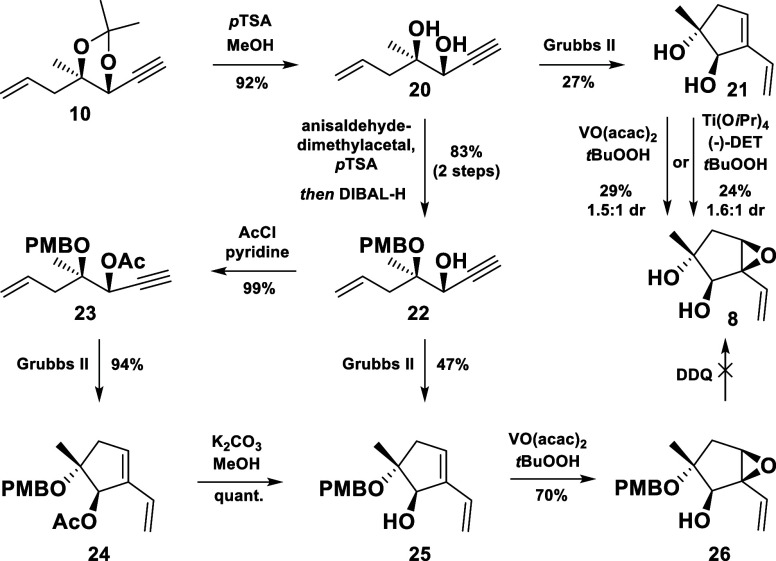 Scheme 4
Scheme 4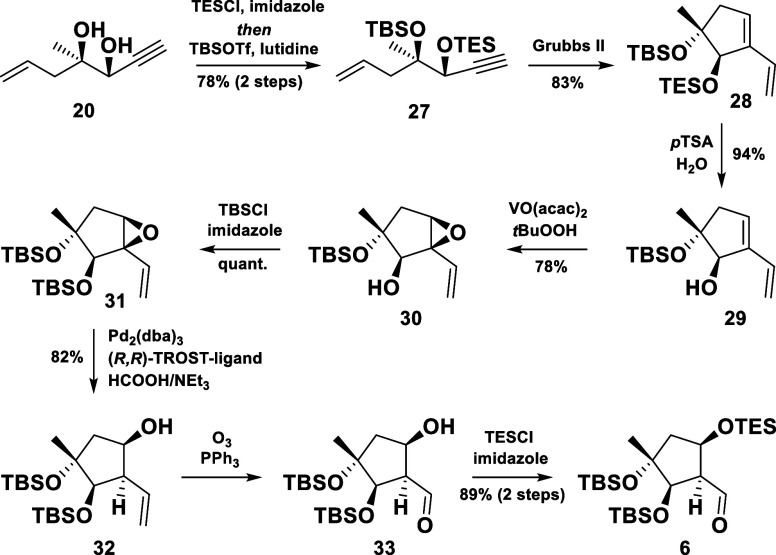 Scheme 5
Scheme 5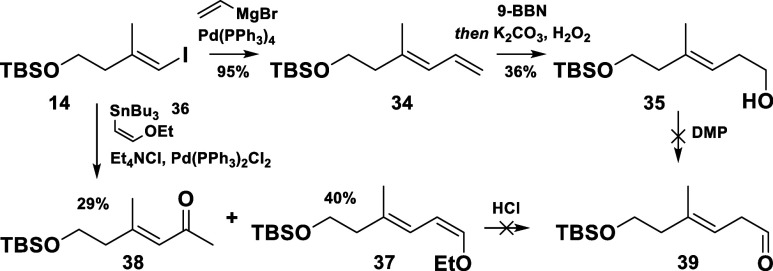 Scheme 6
Scheme 6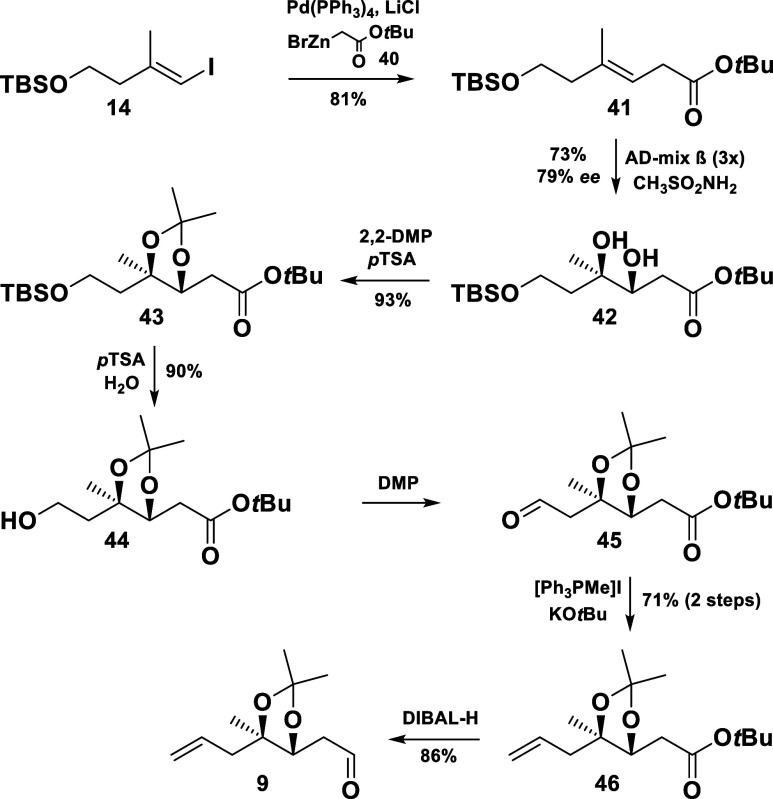 Scheme 7
Scheme 7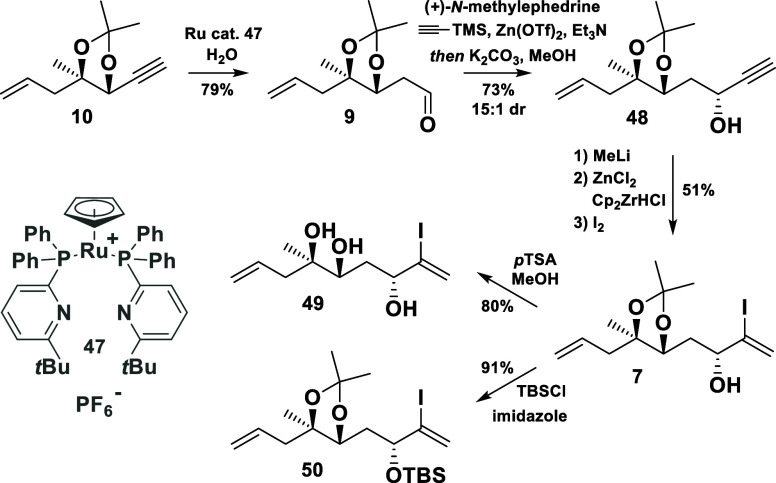 Scheme 8
Scheme 8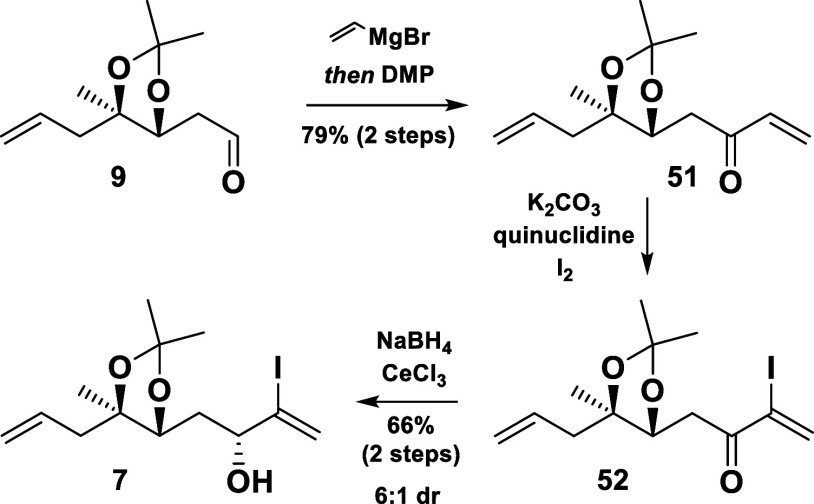 Scheme 9
Scheme 9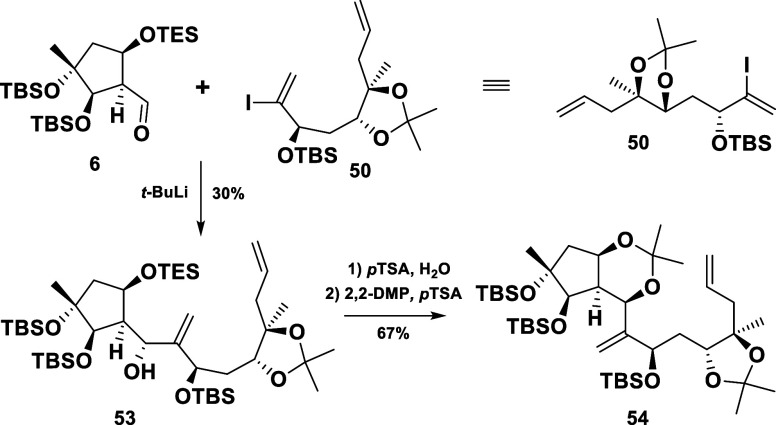 Scheme 10
Scheme 10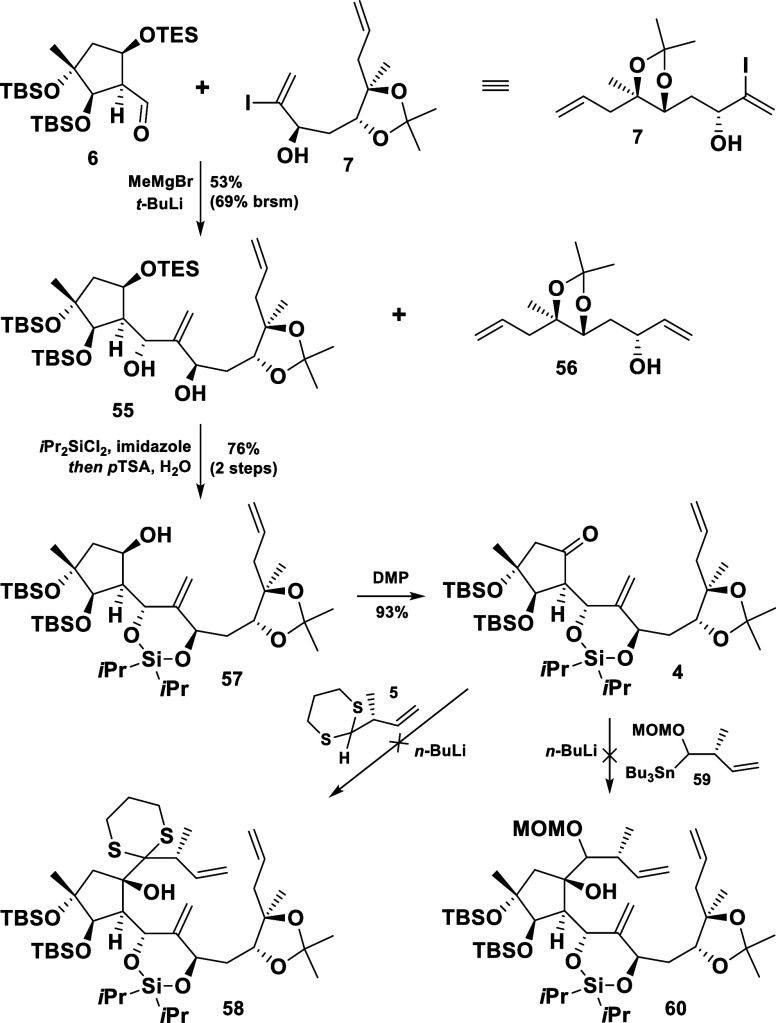 Scheme 11
Scheme 11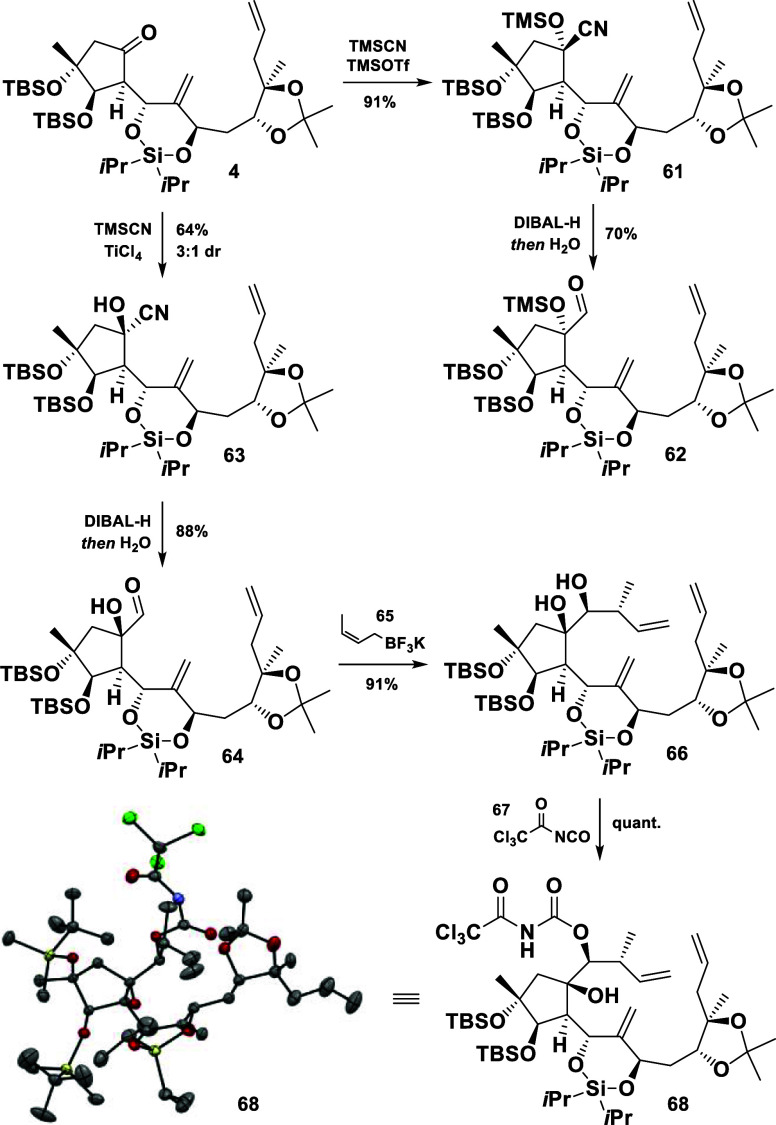 Scheme 12
Scheme 12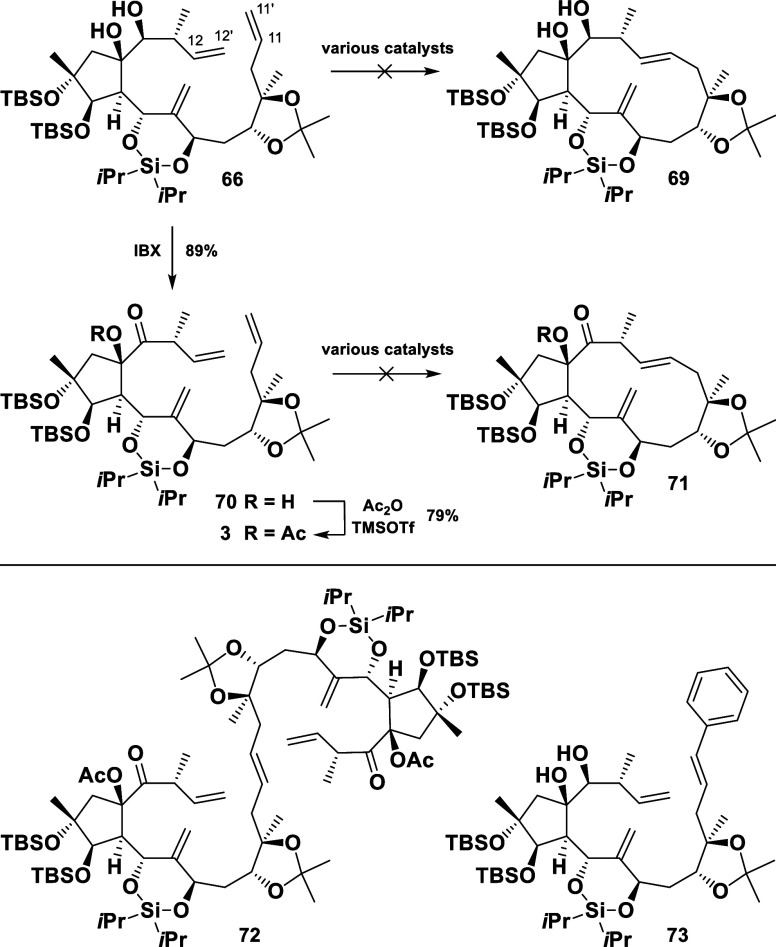 Scheme 13
Scheme 13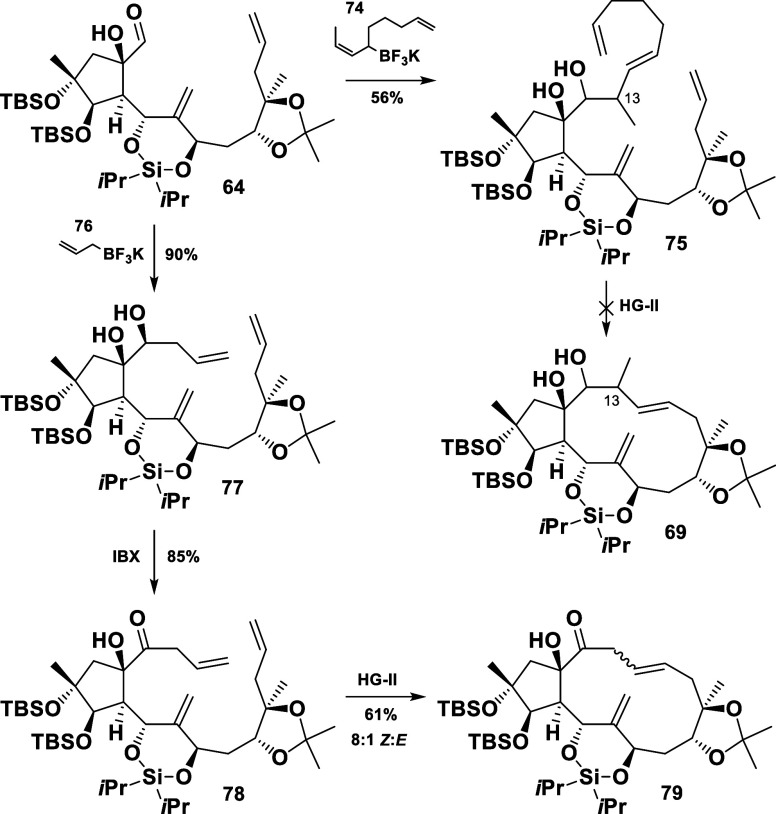 Scheme 14
Scheme 14- —Austrian Science Fund10.13039/501100002428
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSynthesis and Reactivity of Sulfur-Containing Compounds · Marine Sponges and Natural Products · Synthetic Organic Chemistry Methods
Introduction
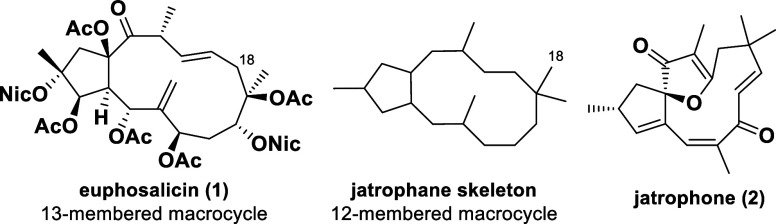
Euphosalicin (1) was first isolated in 2001 by Hohmann et al. from Euphorbia salicifolia, a perennial flowering plant distributed in Central and Southeastern Europe. It is structurally related to the jatrophane diterpenoid family, albeit being considered as the first representative of a new class of bicyclic diterpenes by its discoverers, due to its unique carbon skeleton.^1^
Since the first isolation of jatrophone (2) in 1970 by Kupchan et al.,^2^ interest in this kind of diterpenoids emerged and led to the discovery of numerous jatrophane derivatives.^3^ Many of them display intriguing biological properties, including cytotoxic, antiviral, immunomodulatory, and anti-inflammatory activities. Most notably, a number of jatrophane diterpenes exhibit significant multidrug resistance (MDR) reversal ability.^4^
While syntheses of jatrophone (2) have been reported by Smith, Hegedus, and Wiemer,^5^ synthetic approaches toward other jatrophane diterpenes remain scarce. However, partial syntheses have been described by Yamamura, Mulzer, and Rinner, among others.^6^
A defining feature of jatrophane derivatives is the prevalent bicyclic core, consisting of a cyclopentane motif and an annulated macrocycle. All of the latter ones generally exhibit a 12-membered ring system, whereas the unique 13-membered carbon framework of euphosalicin (1) is surmised to be formed by an incorporation of a geminal methyl group into the ring system.^1^
We have been interested in the synthesis of euphosalicin (1) not only because of the unique and challenging structural motifs (including nine stereocenters and a highly complex oxidation pattern) but also because of its promising biological activities. In their initial studies, Hohmann et al. showed that euphosalicin (1) displays exceptional potential in reversing MDR.^1^
Its remarkable biological properties, coupled with its intricate molecular structure, make 1 an attractive target for its synthetic preparation. Moreover, we envisioned its first total synthesis to facilitate further pharmacological investigations on this outstanding diterpenoid.
Herein, we report our findings en route to the unique bicyclic carbon skeleton of euphosalicin (1) (Scheme 1).
Structures of Euphosalicin (1), Jatrophone (2), and the Jatrophane Carbon SkeletonAc = acetyl, Nic = nicotinoyl.
Our full retrosynthetic strategy is outlined in Scheme 2. The 13-membered macrocycle was envisaged to be prepared via a late stage ring closing metathesis (RCM) of the triene 3, which could be prepared by the addition of the deprotonated dithiane 5 to the ketone 4.^5a,5b^ The synthesis of 4 was planned to be accomplished by the coupling of the aldehyde 6 with the vinyl iodide 7 (after a metal–halogen exchange). This aldehyde 6 was intended to arise from the cyclopentane derivative 8, which in turn could be constructed via a ring-closing enyne metathesis of the intermediate 10.^7^ The enantioselective introduction of the 1,2-diol moiety in 10 was expected to be feasible via a Sharpless dihydroxylation; the required enyne system in 11 was traced back to the commercially available 3-butyn-1-ol (12).
Retrosynthetic Analysis of Euphosalicin (1)
The subunit 7 was envisioned to originate from the aldehyde 9; further simplification led once again to 10 and 3-butyn-1-ol (12), respectively. Thus, the latter would serve as the starting material for both key intermediates 6 and 7.
Results and Discussion
The synthesis commenced with the carboalumination^8^ of 3-butyn-1-ol (12), followed by an iodination giving the (E)-vinyl iodide 13 in 86% yield at decagram scale (Scheme 3). The protection of the free alcohol in 13 and a subsequent Sonogashira coupling afforded the conjugated enyne 11 in excellent yield. Next, a Sharpless dihydroxylation of 11 using a modified AD-mix-β (×3)^9^ gave the desired cis-1,2-diol 15 in 81% yield and 91% enantiomeric excess (ee). The ee of the performed dihydroxylation was determined via^1^H and ^19^F NMR spectra of the corresponding Mosher’s esters, and the absolute (R,R)-configuration was later confirmed by X-ray diffraction measurements of the triol 16 (CCDC no. 2340302).
Synthesis of Compound 10
The synthesis continued with the protection of the diol moiety in 15, followed by a global desilylation to furnish the primary alcohol 18, which was converted to the enyne 10 in two steps.
After the cleavage of the acetonide in 10, the obtained diol 20 was treated with the Grubbs second generation catalyst (Scheme 4).^7^ Even though TLC control showed that the cyclic compound 21 was the main product of the reaction, all attempts to subject the crude reaction mixture to chromatography, in the presence of air, resulted in a substantial loss of material and 21 was isolated in only 27% yield. It has been conceivable that the ruthenium, forming stable complexes with the hydroxyl groups in close proximity, had been responsible for the degradation of the product. The usage of basified cysteine as ruthenium scavenger during work up only marginally enhanced the yield.
RCEYM Studies
However, preliminary experiments on the selective epoxidation of the endocyclic double bond could be carried out with the obtained material. Quite surprisingly, it turned out that the distant homoallylic alcohol had a pronouncedly negative influence on the vanadium catalyzed epoxidation,^7^ partially directing the catalyst toward the other face of the molecule. Consequently, a mixture of both epoxides was produced with a diastereomeric ratio of 1.5:1. An attempt to improve the selectivity by a Sharpless asymmetric epoxidation only resulted in a slight diastereomeric excess of the desired epoxide 8 once more. At this point it was anticipated that the selective protection of the tertiary alcohol could serve its purpose in both, in optimizing the yield of the RCEYM, as well as in improving the diastereomeric ratio for the desired epoxide 8 upon epoxidation. The PMB protected derivative 22 was accessed through an acetal formation and a regioselective reductive opening in 83% yield and subjected to an RCEYM (Scheme 4). While the yield of the cyclopentane 25 improved to 47%, it was still not satisfying. To test the hypothesis of free hydroxyl groups being an issue, a fully protected 1,2-diol 23 was prepared, which finally gave the cyclized product 24 in an excellent yield. Even though this synthetic path could be considered a detour, it provided the desired cyclic allylic alcohol 25 in a significantly improved overall yield (93%) after deacetylation. With the homoallylic alcohol masked, the vanadium catalyzed epoxidation smoothly furnished the desired epoxide 26 in good yield as a single diastereomer. Following our synthetic plan, silyl ethers were to be installed to protect the 1,2-diol moiety, which required the PMB group to be removed. Unfortunately, all endeavors to execute this transformation failed.
With these lessons in mind, a slightly modified approach was pursued. Hence, the selective installation of TES and TBS ethers onto the diol 20, followed by an RCEYM and the removal of the TES group, ultimately afforded 29 in 62% yield over four steps (Scheme 5). The previously discussed epoxidation method then yielded 30 as the sole product in 78% yield. At this stage, the stereochemical outcome of the epoxidation was proven via X-ray single crystal diffraction of 30 (CCDC no. 2340303). With an efficient and reliable access to the epoxide 30 secured, we targeted the final steps toward the desired cyclopentane motif.
Synthesis of the Cyclopentane Motif
Thus, the secondary alcohol was smoothly protected as its TBS ether, and the subsequent reductive opening of the allylic epoxide 31 was carried out following a protocol developed by Rinner et al. (Scheme 5).^7^ Using their optimized conditions, the desired secondary alcohol 32 was isolated as a single diastereomer in 82% yield. For the remaining two steps toward the first building block, the vinyl group was ozonolyzed to give the β-hydroxy aldehyde 33. To our delight, X-ray diffraction measurements of this crystalline material confirmed the depicted stereochemistry (CCDC no. 2340304). Finally, a TES protection of the hydroxyl group completed the synthesis of the cyclopentane fragment 6.
Next, the synthesis of the vinyl iodide fragment 7 was tackled. Four different approaches toward the synthesis of the aldehyde 9 (the obvious precursor for the vinyl iodide 7) were tested. They were running in parallel with the idea to push forward with the most promising one to completion. The attempts commenced with a Kumada coupling of the vinyl iodide 14 to furnish the diene 34 (Scheme 6).^10^ Unfortunately, the subsequent hydroboration-oxidation sequence resulted in a very low yield and the DMP oxidation of resulting primary alcohol 35 led to a partial double bond migration.
Kumada- and Stille Coupling Approaches toward the Aldehyde 39
The second approach included a palladium-mediated cross coupling between 14 and the vinyl stannane 36 to afford the desired compound 37 in a moderate yield.^11^ A competitive Heck-type reaction with ethyl vinyl ether, presumably formed by the decomposition of the tin-organic reagent, was elucidated as the main cause for the diminished yield, resulting in the coformation of the ketone 38. Regrettably, the attempts to hydrolyze the obtained enol ether 37 resulted in an inseparable mixture of the target aldehyde 39 and its isomerized conjugated product (not shown in the scheme).
In our third route (Scheme 7), the vinyl iodide 14 was converted into the tert-butyl ester 41,^12^ which was subjected to a dihydroxylation to give the 1,2-diol 42 in 73% yield and 79% ee (determined by ^1^H and ^19^F NMR spectra of the corresponding Mosher’s esters). Pleasantly, we could rely on our developed protocols for the following protecting group manipulations and the subsequent transformations. Only the procedure for the cleavage of the TBS ether had to be changed, as the basicity of TBAF triggered an elimination through enolization of the tert-butyl ester 43. Acidic conditions, however, smoothly furnished the desired primary alcohol 44. In analogy to the preparation of the enyne 10, the synthesis proceeded with the previously developed oxidation-olefination sequence to access the olefin 46. Gratifyingly, the key aldehyde intermediate 9 could be obtained by the reduction of the ester 46 at low temperature.
Negishi Coupling Approach toward the Aldehyde 9
In terms of the last approach toward 9, the hydration of the terminal triple bond in 10 was carried out utilizing a ruthenium catalyst to give the identical aldehyde 9 in 79% yield (Scheme 8).^13^ It should be noted that, although both successful routes to access 9 (Schemes 7, 3, and 8) were comparable in terms of the number of steps and the overall yield (from 14), the enantiopurity in the last one was found to be much better (91 vs 79% ee). This was our main criterion to continue with the hydration approach, leaving the other one as a backup.
Synthesis of the Vinyl Iodide Building Block 7
With compound 9 in hand, the synthesis of 7 was successfully completed (Scheme 8). Following Carreira’s protocol^14^ for the enantioselective alkynylation of aldehydes, the alkyne 48 was prepared in 73% yield and 88% de. Next, the hydrometalation of the terminal triple bond was attempted. Whereas hydroalumination or hydrosilylation protocols failed to deliver the desired regioisomer,^15^ a hydrozirconation procedure for propargylic alcohols, developed by Zhang and Ready,^16^ readily gave the α-vinyl iodide 7 as a single isomer in 51% yield.
The expected stereochemical outcome of the enantioselective alkyne addition was proven by X-ray diffraction measurement of the triol 49, which could be obtained after the cleavage of the acetal group (CCDC no. 2340305). Additionally, its protected derivative 50 was synthesized, even though there were substantial concerns regarding the potential tendency of the corresponding metalated species to collapse into an allene (not shown in the scheme). It should be noted that the described conversion of 48 to the vinyl iodide 7 was very capricious. The yield strongly depended on the quality of the reagents and the scale of the reaction.
Thus, a reproducible and more reliable synthesis of 7 was developed later on (Scheme 9). Beginning anew with the aldehyde 9, a vinyl group was added, followed by an oxidation to afford the α,β-unsaturated ketone 51. After employing a Baylis–Hillman-type iodination protocol,^17^ a Luche reduction was executed to deliver the α-vinyl iodide 7 in good yield (52% from 9) and diastereoselectivity. Additionally, the undesired diastereomer (not shown in the scheme) could be recycled via oxidation.
Alternative Approach toward 7
With both building blocks (6 and 7) in hand, the stage was set for their coupling to obtain compound 55via a nucleophilic addition. At the beginning, however, we decided to utilize the protected vinyl iodide 50 for the coupling. This choice was expected to allow initial insights in both stereoselectivity and reactivity. Furthermore, it could facilitate the inversion of the newly formed chiral center without selectivity problems, if needed. The subjection of the vinyl iodide 50 to t-BuLi and its subsequent addition to the aldehyde 6 furnished the addition product 53 as a single diastereomer in 30% yield (Scheme 10). To elucidate the stereochemistry of the newly formed chiral center, compound 53 was converted to the rigid bisketal 54. Gratifyingly, NOESY supported investigations confirmed the desired stereochemical outcome of the addition reaction (see Supporting Information).
Initial Attempts at Fragment Coupling
The low yield of the described reaction, most probably associated with the instability of the organolithium reagent (α-elimination of the OTBS group) prompted us to use the unprotected vinyl iodide 7 in this crucial coupling (Scheme 11). This proved to be beneficial, as the lithiation of the preformed Mg salt of 7 (MeMgBr, −10 °C) and its subsequent coupling with the aldehyde 6 afforded the 1,3-diol 55 in 53% yield. Again, the remarkable stereocontrol by the aldehyde substrate could be observed and the coupling product 55 was isolated as a single diastereomer. Some unreacted starting material 6 was recoverable and the dehalogenated side product 56 could be recycled, which emphasized the superiority of the developed alternative approach toward the vinyl iodide 7 (Scheme 9).
Proceedings toward the Cyclopentanone 4 and Failed Alkylations
Guided by the initial plan, the 1,3-diol moiety in intermediate 55 was protected as a cyclic silyl ether before the TES group was removed selectively, to furnish 57. A subsequent oxidation afforded the ketone 4, which was the starting point for the installation of the C12–C14/C20 (northern) fragment via the addition of the lithiated 1,3-dithiane 5(18) or the coupling with 59 as an alternative (after a Sn/Li exchange).^19^ Unfortunately, the attempted alkylations failed under a variety of conditions (altering temperature and reaction time, addition of CeCl_3_ and LaCl_3_).^20^ Instead, extensive decomposition, epimerization, and eliminations of the ketone 4 were observed, most probably due to enolate formation and the steric hindrance in the vicinity of the carbonyl group.
Based on these observations, the formation of a cyanohydrin 63 was proposed, arguing that a cyanide anion might be small enough to overcome the steric obstruction. Indeed, TMSOTf-mediated cyanohydrin formation conditions smoothly furnished the TMS-protected product 61 as a single diastereomer (Scheme 12).^21^ After a reduction to deliver the aldehyde 62, we were surprised to undoubtedly identify the latter as the depicted, undesired diastereomer (NOESY correlations, Supporting Information). It was speculated that the reversibility of the cyanohydrin formation may be responsible for that counterintuitive outcome, favoring the thermodynamic product 61. To avoid this problem, a more oxophilic Lewis acid was employed to potentially retard the reversibility. Indeed, a TiCl_4_-mediated addition preferentially gave the desired cyanohydrin 63, confirmed by NOESY correlations of the corresponding aldehyde 64 (see Supporting Information).^22^
Synthesis of 66 and Its Derivatization for X-ray Measurement
The synthesis continued with a crotylation to introduce the remaining carbon atoms of the northern fragment (Scheme 12). Whereas a Roush crotylation failed,^23^ zinc or indium promoted reactions were unfortunately not selective and afforded 66 as a mixture of diastereomers (not shown in the scheme). In contrast, the treatment of the aldehyde 64 with the crotyltrifluoroborate 65 cleanly furnished the desired 1,2-diol 66 as a single product in 91% yield.^24^ The remarkable substrate control may originate from a fixed arrangement of the aldehyde group through hydrogen bonding with the adjacent tertiary hydroxyl group, whereas the syn selectivity was achieved by employing the (Z)-crotyltrifluoroborate. The stereochemistry of the afforded product 66 was unambiguously confirmed by X-ray diffraction measurements of the carbamate derivative 68 (CCDC no. 2337864).
The stage was now set for the key macrocyclization of the triene 66 by means of an RCM reaction. Unfortunately, all endeavors to execute this key transformation were ultimately unsuccessful. Neither the ketone 70 nor its acetylated derivative 3 underwent an RCM (Scheme 13). To this end, a variety of metathesis catalysts had been assessed, including first and second generations of Grubbs and Grubbs–Hoveyda catalysts. Furthermore, very active, less commonly employed catalysts like Nitro-Grela or Grubbs third generation as well as a Schrock molybdenum catalyst failed to deliver the desired products (see Supporting Information).^25^ Additionally, the substitution of the cyclic silyl ether with acetyl groups for increased flexibility did not facilitate the cyclization (not shown in the scheme).
Failed Macrocyclization Attempts
It was observed, that in all attempts where the starting material underwent conversion (high catalyst loadings and long reaction times), only the eastern (11/11′) double bond was addressed by the ruthenium, resulting in either a dimer formation at this position (72) or in a regioselective cross metathesis with the catalyst (Grubbs II) itself (73). The experimental details of the RCM investigations can be found in the Supporting Information.
It was decided to evaluate a relay metathesis approach to force the catalyst to incorporate the northern (12/12′) double bond into the reaction.^26^ Accordingly, the necessary moiety was installed by treating the aldehyde 64 with the modified trifluoroborate salt 74 (see Supporting Information) to give the compound 75 in 56% yield (Scheme 14). Regrettably, the efforts only resulted in the cleavage of the tether and no macrocyclization occurred.^27^
Studies toward the Successful Macrocyclization
It seemed that the steric hindrance around the methyl group at position 13 was preventing the macrocyclization and could not be overcome, not even via a relay metathesis (Scheme 14). To corroborate this assumption, the aldehyde 64 was elaborated to the modified triene 78, now lacking the notorious methyl group. Finally, we succeeded in isolating the macrocycle 79, even though the RCM had favored the formation of the undesired (Z) geometry of the double bond. Unfortunately, its isomerization failed under a variety of conditions (iodine mediated, UV irradiation, AIBN/PhSH) and no conversion to the desired (E) derivative was observed.^5a,5c,28^ Nevertheless, the studies on the macrocyclization culminated in the successful isolation of the C11/C12 (Z) isomer of the C13 nor methyl skeleton of euphosalicin (1).
Conclusions
Although the targeted first total synthesis of euphosalicin (1) was unsuccessful, we accomplished the stereoselective synthesis of the seco compound 3 with all nine stereocenters installed in correct manner. The successful macrocyclization of 78 afforded the C11/C12 (Z) isomer of the C13 nor methyl skeleton of euphosalicin (1). We are confident that the unique findings en routeto synthesize the natural product 1 presented herein provide invaluable insights for future attempts toward the synthesis of 1 in particular and jatrophane diterpenoids in general. Additionally, detailed studies on RCEYM and macrocyclizations should aid researchers to gain a deeper understanding of these important transformations in total synthesis.
Experimental Section
Compound 13
To a stirred suspension of Cp_2_ZrCl_2_ (2.75 g, 9.4 mmol, 0.22 equiv) in dry DCM (120 mL) in a Schlenk flask, AlMe_3_ (2 M in toluene, 64 mL, 128 mmol, 3 equiv) was added via a syringe at −25 °C. The resulting yellow mixture was stirred at −25 °C for 15 min. After dropwise addition of deion. water (1.23 mL, 68.5 mmol, 1.6 equiv), the reaction was stirred again for 20 min at respective temperature. Then, 3-butyn-1-ol 12 (3 g, 42.8 mmol, 1 equiv), pretreated with AlMe_3_ (2 M in toluene, 6.42 mL, 12.8 mmol, 0.3 equiv) in dry DCM (30 mL) at 0 °C, was added via a syringe. The reaction was allowed to reach room temperature and was stirred overnight.
The resulting yellow slurry was again cooled to −25 °C, and a solution of I_2_ (21.7 g, 85.6 mmol, 2 equiv) in dry diethyl ether (150 mL) was added via a syringe. The mixture was allowed to reach room temperature and stirred for 4 h. The reaction was quenched by the addition of 40 mL sat. Na–K-tartrate-solution and stirred until the aluminum was fully complexed. The organic phase was decanted off, and the precipitate was washed several times with diethyl ether. The combined organic phases were washed once with sat. Na_2_S_2_O_3_ solution and once with brine, dried over Na_2_SO_4_, filtered, and concentrated. The crude product was purified via column chromatography (petroleum ether/ethyl acetate, 5:1) to afford 7.81 g (86%) of the title compound 13 as brown oil. ^1^H NMR (400 MHz, CDCl_3_): δ 6.02 (q, J = 1.1 Hz, 1H), 3.72 (t, J = 6.3 Hz, 2H), 2.48 (td, J = 6.3, 1.1 Hz, 2H), 1.87 (d, J = 1.1 Hz, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 144.9, 77.7, 60.4, 42.7, 24.1. HRMS (ESI) m/z: [M + H]^+^ calcd for C_5_H_10_IO, 212.9771; found, 212.9774.
Physical and spectral data were in accordance with the literature.^29^
Compound 14
To a stirred solution of 13 (10 g, 47.2 mmol, 1 equiv) in DCM (200 mL), imidazole (8 g, 117.9 mmol, 2.5 equiv) and chloro tert-butyldimethylsilane (8.5 g, 56.6 mmol, 1.2 equiv) were added. The reaction was stirred for 1 h until TLC had indicated complete conversion. The mixture was then quenched by the addition of water. The aqueous phase was extracted thrice with DCM, and the combined organic phases were washed with brine, dried over Na_2_SO_4_, and concentrated. The residue was purified via flash chromatography (petroleum ether/ethyl acetate, 70:1) to yield 14.6 g (95%) of the TBS-protected alcohol 14 as yellow oil. ^1^H NMR (400 MHz, CDCl_3_): δ 5.93 (h, J = 1.1 Hz, 1H), 3.68 (t, J = 6.6 Hz, 2H), 2.41 (td, J = 6.6, 1.1 Hz, 2H), 1.85 (d, J = 1.1 Hz, 3H), 0.88 (s, 9H), 0.04 (s, 6H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 145.3, 76.5, 61.5, 42.7, 26.0, 24.4, 18.4, −5.2. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_11_H_23_IOSiNa, 349.0455; found, 349.0448.
Physical and spectral data were in accordance with the literature.^30^
Compound 11
To a solution of 14 (22 g, 67.4 mmol, 1 equiv) in Et_2_NH (500 mL), PdCl_2_(PPh_3_)2 (236 mg, 0.34 mmol, 0.01 equiv) and CuI (2.57 g, 13.5 mmol, 0.2 equiv) were added. The reaction mixture was stirred under light protection for 10 min at 10 °C. After the addition of TMS-acetylene (7.28 g, 10.56 mL, 74.2 mmol, 1.1 equiv) at 10 °C, the reaction was allowed to reach room temperature and stirred for 1 h. After TLC had indicated complete conversion, the reaction was quenched by the addition of sat. NH_4_Cl solution; the organic compound was extracted three times with Et_2_O, and the combined organic phases were dried over Na_2_SO_4_, filtered, and concentrated under reduced pressure. The crude product was purified via flash column chromatography (petroleum ether/ethyl acetate, 70:1) to yield 18.12 g of 11 (91%) as yellow oil. Alternatively, the product can be purified via Kugelrohr distillation. (0.5 mbar, 110 °C) ^1^H NMR (400 MHz, CDCl_3_): δ 5.33 (q, J = 1.2 Hz, 1H), 3.68 (t, J = 6.9 Hz, 2H), 2.29 (td, J = 6.9, 1.2 Hz, 2H), 1.93 (d, J = 1.2 Hz, 3H), 0.88 (s, 9H), 0.19 (s, 9H), 0.04 (s, 6H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 151.1, 106.7, 103.4, 97.1, 61.9, 42.1, 26.1, 20.1, 18.4, 0.3, −5.2. HRMS (ESI) m/z: [M – H]^−^ calcd for C_16_H_31_OSi_2_, 295.1919; found, 295.1922.
Compound 15
Potassiumosmate dihydrate (220 mg, 600 μmol) and (DHQD)2_PHAL (2.34 g, 3 mmol) were added to a mixture of powdered K_3_Fe(CN)6 (98 g, 300 mmol) and K_2_CO_3 (41.2 g, 300 mmol). The resulting mixture was ground to afford 141.8 g of AD-mix-β with 3x increased osmate concentration.
To a mechanically stirred suspension of AD-mix-β-(3×) (118 g, 1.4 g/mmol) in t-BuOH/H_2_O (100 mL each) was added methanesulfonamide (24 g, 252.8 mmol, 3 equiv). After 2 h of stirring, the mixture was cooled to 0 °C before compound 11 (25 g, 84.3 mmol, 1 equiv) was added. The orange suspension was then stirred for 6 days at 0 °C until TLC had indicated complete conversion. During this period, the color of the reaction mixture gradually changed from orange to yellow. The reaction was quenched with solid Na_2_SO_3_, causing a color change to gray and allowed to reach room temperature. Ether was added, and the mixture was stirred for 30 min. The organic compound was extracted five times with ether and the combined organic phases were dried over Na_2_SO_4_, filtered, and concentrated to obtain a crude product which was purified via column chromatography (petroleum ether/diethyl ether, 1:1) to yield 22.6 g (81%) of the diol 15 as colorless oil. ^1^H NMR (400 MHz, CDCl_3_): δ 4.26 (d, J = 5.0 Hz, 1H), 3.89 (qdd, J = 10.8, 7.0, 4.2 Hz, 2H), 3.82 (s, 1H), 3.19 (d, J = 4.9 Hz, 1H), 1.94–1.77 (m, 2H), 0.90 (s, 9H), 0.16 (s, 9H), 0.09 (s, 3H), 0.09 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 103.8, 91.2, 75.1, 69.8, 60.3, 39.3, 25.9, 22.2, 18.2, 0.0, −5.5. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_16_H_34_O_3_Si_2_Na, 353.1938; found, 353.1941. Specific rotation: [α]D^20^ +11.8 (c 1.00, CH_2_Cl_2_).
Compound 16
To a solution of 15 (100 mg, 302 μmol, 1 equiv) in THF (3 mL) and H_2_O (400 μL) was added p-toluenesulfonic acid (5 mg, 30 μmol, 0.1 equiv). The mixture was then stirred at room temperature until TLC had indicated full conversion (24 h). Subsequently, saturated aqueous NaHCO_3_ solution was added and the aqueous phase was extracted with ether. The combined organic layers were washed with H_2_O and brine, dried over Na_2_SO_4_, filtered, and concentrated to give 60 mg (92%) of triol 16 as white crystals. ^1^H NMR (400 MHz, CDCl_3_): δ 4.27 (s, 1H), 3.95 (ddd, J = 11.5, 7.9, 3.7 Hz, 1H), 3.86 (ddd, J = 11.0, 6.7, 4.0 Hz, 1H), 2.94 (s, 1H), 2.70 (s, 1H), 2.57 (s, 1H), 1.95 (ddd, J = 14.8, 8.0, 4.0 Hz, 1H), 1.82 (ddd, J = 14.8, 6.7, 3.7 Hz, 1H), 1.34 (s, 3H), 0.18 (s, 9H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 103.5, 92.1, 75.8, 69.8, 59.5, 39.0, 22.3, −0.1. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_10_H_20_O_3_SiNa, 239.1074; found, 239.1071. Specific rotation: [α]D^20^ +18.0 (c 1.00, CH_2_Cl_2_). Melting point: mp 88.7–89.8 °C.
Compound 17
To a stirred mixture of 15 (20 g, 60.5 mmol, 1 equiv) and molecular sieve (4 Å) in dry DCM (500 mL) were added p-toluenesulfonic acid (1 g, 6.1 mmol, 0.1 equiv) and 2,2-dimethoxypropane (18.9 g, 22.2 mL, 181.5 mmol, 3 equiv) at 0 °C. The resulting suspension was stirred for 5 h at respective temperature. Once TLC had indicated full completion, the reaction was quenched with sat. NaHCO_3_ solution. The whole mixture was then filtered over Celite, before the product was extracted several times with DCM. The combined organic phases were dried over Na_2_SO_4_, filtered, and concentrated. The crude product was purified via column chromatography (petroleum ether/ethyl acetate, 15:1) to obtain 19.8 g (88%) of the acetal protected product 17 as colorless oil. ^1^H NMR (400 MHz, CDCl_3_): δ 4.74 (s, 1H), 3.83–3.69 (m, 2H), 1.84 (td, J = 6.7, 3.5 Hz, 2H), 1.49 (s, 3H), 1.34 (s, 3H), 1.32 (s, 3H), 0.90 (s, 9H), 0.17 (s, 9H), 0.06 (s, 6H), 0.06 (s, 6H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 108.9, 100.8, 93.1, 82.4, 73.6, 59.1, 41.8, 28.5, 27.2, 26.0, 23.4, 18.3, −0.1, −5.2, −5.3. HRMS (ESI) m/z: [M – H]^−^ calcd for C_19_H_37_O_3_Si_2_, 369.2286; found, 369.2282. Specific rotation: [α]D^20^ +13.4 (c 1.00, CH_2_Cl_2_).
Compound 18
A solution of 17 (15 g, 40.5 mmol, 1 equiv) in dry THF (400 mL) was chilled to 0 °C. Subsequently, tetrabutylammonium fluoride (1 M in THF, 89 mL, 89.0 mmol, 2.2 equiv) was added via a syringe. The resulting dark brown solution was allowed to reach room temperature and stirred for 2 h until TLC had indicated full conversion. The reaction mixture was then quenched by the addition of sat. NH_4_Cl solution, causing a color change to yellow. The organic compound was extracted three times with ether, and the combined organic phases were dried over Na_2_SO_4_, filtered, and concentrated under reduced pressure. The crude product was purified via flash column chromatography (ether/petroleum ether, 2:1) to yield 6.74 g of 18 (90%) as yellowish oil. ^1^H NMR (400 MHz, CDCl_3_): δ 4.57 (d, J = 2.2 Hz, 1H), 3.93–3.74 (m, 2H), 2.55 (d, J = 2.2 Hz, 1H), 2.46 (dd, J = 6.1, 5.0 Hz, 1H), 1.88 (t, J = 5.7 Hz, 2H), 1.50 (s, 3H), 1.38 (s, 6H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 109.6, 83.6, 78.5, 76.4, 73.5, 59.2, 40.3, 28.4, 27.1, 22.8. HRMS (ESI) m/z: [M – H]^−^ calcd for C_10_H_15_O_3_, 183.1026; found, 183.1015. Specific rotation: [α]D^20^ +9.9 (c 1.00, CH_2_Cl_2_).
Compound 19
To a stirred solution of the primary alcohol 18 (4 g, 21.7 mmol, 1 equiv) in DCM (200 mL) were added solid NaHCO_3_ (5.5 g, 65.1 mmol, 3 equiv) and Dess-Martin periodinane (11.1 g, 26.1 mmol, 1.2 equiv) at room temperature. The reaction mixture slightly warmed up and was stirred until TLC had indicated full conversion (30 min). The suspension was then directly filtered over silica (100 g) and eluted with DCM. The product containing fractions were combined, and DCM was distilled off (40 °C, 700 mbar) to give the crude aldehyde 19 as a volatile, colorless liquid. Due to the volatility of 19, great caution was required during the removal of DCM. It was not necessary to remove the DCM completely, as it does not cause problems in the next reaction.
The obtained crude material was directly used for the next step without further purification. However, an analytical sample was purified via column chromatography (DCM), to collect NMR spectra and physical data. ^1^H NMR (400 MHz, CDCl_3_): δ 9.85–9.82 (t, J = 2.7 Hz, 1H), 4.60 (d, J = 2.2 Hz, 1H), 2.65 (d, J = 2.7 Hz, 2H), 2.59 (d, J = 2.2 Hz, 1H), 1.52 (s, 3H), 1.46 (s, 3H), 1.37 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 200.5, 110.4, 81.1, 78.2, 77.1, 73.4, 52.0, 28.4, 27.2, 23.6. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_10_H_14_O_3_Na, 205.0835; found, 205.0836. Specific rotation: [α]D^20^ +21.3 (c 1.00, CH_2_Cl_2_).
Compound 10
To a stirred suspension of methyltriphenylphosphonium iodide (13.2 g, 32.6 mmol, 1.5 equiv), which was dried by coevaporation with toluene before use, in dry ether (150 mL) at 0 °C was added KOtBu (3.2 g, 28.3 mmol, 1.3 equiv). After stirring the resulting orange suspension for 45 min at the same temperature, a solution of the aldehyde 19 (3.96 g, 21.7 mmol, 1 equiv) in dry ether (50 mL) was added via a syringe.
The mixture was slowly warmed up to room temperature while precipitation occurred. After the reaction had been stirred for 30 min, TLC indicated complete conversion. The reaction was quenched with sat. NH_4_Cl solution and extracted twice with Et_2_O. The combined organic layers were washed once with water and brine, dried over Na_2_SO_4_, filtered, and concentrated (50 °C, ambient pressure). The residue was chromatographed on silica gel (pentane/ether, 15:1) to provide 3.08 g (79% over 2 steps) of the olefin 10 as a colorless, volatile liquid.
Due to the volatility of the olefin 10, pentane and ether were carefully distilled off at 50 °C at ambient pressure. ^1^H NMR (400 MHz, CDCl_3_): δ 5.86 (ddt, J = 16.8, 10.4, 7.3 Hz, 1H), 5.24–5.06 (m, 2H), 4.52 (d, J = 2.2 Hz, 1H), 2.54 (d, J = 2.2 Hz, 1H), 2.43–2.31 (m, 2H), 1.50 (s, 3H), 1.36 (s, 3H), 1.34 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 133.3, 118.8, 109.4, 82.9, 79.3, 76.2, 72.5, 43.8, 28.5, 27.3, 23.1. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_11_H_16_O_2_Na, 203.1042; found, 203.1037. Specific rotation: [α]D^20^ +17.2 (c 1.00, CH_2_Cl_2_).
Compound 20
To a solution of 10 (3 g, 16.6 mmol, 1 equiv) in MeOH (160 mL), p-toluenesulfonic acid (573 mg, 3.3 mmol, 0.2 equiv) was added in one portion. The resulting mixture was heated up to 50 °C (oil bath) and stirred for 24 h at respective temperature.
After TLC had indicated complete conversion, the solvent was removed under reduced pressure and the residue was chromatographed on silica gel (petroleum ether/ethyl acetate, 2:1) to yield 2.15 g (92%) of the diol 20 as colorless oil. ^1^H NMR (400 MHz, CDCl_3_): δ 6.00–5.75 (m, 1H), 5.24–5.03 (m, 2H), 4.21 (dd, J = 6.3, 2.2 Hz, 1H), 2.51 (d, J = 2.2 Hz, 1H), 2.43–2.36 (m, 3H), 2.07 (s, 1H), 1.30 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 132.9, 119.4, 81.9, 74.9, 74.4, 68.6, 42.3, 22.2. HRMS (ESI) m/z: [M + H]^+^ calcd for C_8_H_13_O_2_, 141.0910; found, 141.0913. Specific rotation: [α]D^20^ +10.6 (c 1.00, CH_2_Cl_2_).
Compound 21
The starting material 20 (80 mg, 571 μmol, 1 equiv) was dissolved in dry ethyl acetate (50 mL) before the reaction mixture was degassed via freeze–pump–thaw cycles (3×). After addition of the Grubbs second generation catalyst [246047-72-3] (24 mg, 28 μmol, 0.05 equiv), an ethylene atmosphere was created, which was maintained throughout the reaction. The slightly pink homogeneous solution was stirred overnight at 55 °C (oil bath). As TLC had indicated incomplete conversion, another 2 mol % of the catalyst (9 mg) was added and the reaction was stirred for 5 h under ethylene atmosphere. Next, it was exposed to air to oxidize the remaining catalyst. The reaction mixture was then filtered over a short plug of silica, washed out with ether, before the solvents were distilled off. Crude brown oil was obtained, which was purified via column chromatography (petroleum ether/ethyl acetate, 2:1) to yield 22 mg (27%) of the cyclopentane 21. ^1^H NMR (400 MHz, CDCl_3_): δ 6.42 (dd, J = 17.7, 10.9 Hz, 1H), 5.84 (t, J = 2.8 Hz, 1H), 5.43 (d, J = 17.7 Hz, 1H), 5.17 (d, J = 10.4 Hz, 1H), 4.48 (s, 1H), 2.67–2.51 (m, 1H), 2.45–2.37 (m, 1H), 2.10 (s, 1H), 2.03 (s, 1H), 1.40 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 143.3, 131.9, 131.7, 115.8, 82.8, 81.1, 45.9, 22.5. HRMS (ESI) m/z: [M – H]^−^ calcd for C_8_H_11_O_2_, 139.0764; found, 139.0762. Specific rotation: [α]D^20^ +7.9 (c 1.00, CH_2_Cl_2_).
Compound 8
Method 1: to a solution of 21 (70 mg, 499 μmol, 1 equiv) in dry DCM (5 mL) at 0 °C was added VO(acac)2 (26 mg, 100 μmol, 0.2 equiv) in one portion, followed by the dropwise addition of tert-butylhydroperoxide (5.5 M in decane, 100 μL, 549 μmol, 1.1 equiv). The resulting red solution was allowed to reach room temperature. After being stirred for 1 h, TLC had indicated complete conversion. The reaction was quenched by the addition of a saturated aqueous solution of Na_2_S_2_O_3_ and a saturated aqueous solution of NH_4_Cl. The aqueous layer was extracted with DCM, the combined organic layers were dried over Na_2_SO_4_, filtered, and reduced in vacuo. The residue was purified by column chromatography (petroleum ether/ether, 2:1) to give 23 mg (29%) of the epoxide 8 as colorless oil.
Method 2: a Schlenk flask, charged with dry DCM (5 mL) and molecular sieves was placed in a cooling bath (−20 °C). Then (−)-DET (0.2 M in DCM, 500 μL, 100 μmol, 0.2 equiv) and Ti(OiPr)4 (0.2 M in DCM, 375 μL, 75 μmol, 0.15 equiv) were added via a syringe and the reaction was stirred for 15 min at −20 °C. After the dropwise addition of tert-butylhydroperoxide (5.5 M in decane, 90 μL, 499 μmol, 1 equiv), the reaction was stirred for 40 min at the respective temperature. Subsequently, the diol 21 (70 mg, 499 μmol, 1 equiv) was added in dry DCM (1 mL). The resulting mixture was allowed to reach room temperature and stirred overnight. As the reaction was not finished, 0.3 equiv of t-BuOOH was added at −20 °C. Again, the reaction was allowed to reach room temperature and stirred for another 12 h. As soon as TLC had indicated complete conversion, the reaction was quenched with 30% NaOH solution saturated with NaCl at −10 °C and stirred at room temperature for 45 min (slightly orange suspension). The mixture was filtered over a short plug of Celite, dried over Na_2_SO_4_, filtered, and concentrated. The crude product was purified via column chromatography (petroleum ether/ethyl acetate, 2:1) to give 19 mg (24%) of the epoxide 8 as yellowish oil. ^1^H NMR (600 MHz, CD_2_Cl_2_): δ 6.08 (dd, J = 17.5, 11.0 Hz, 1H), 5.41 (dd, J = 17.5, 1.6 Hz, 1H), 5.25 (dd, J = 11.0, 1.6 Hz, 1H), 3.69 (s, 1H), 3.49 (q, J = 0.9 Hz, 1H), 3.04 (s, 1H), 2.86 (s, 1H), 1.95 (dd, J = 15.0, 1.1 Hz, 1H), 1.91 (dd, J = 14.9, 0.8 Hz, 1H), 1.14 (s, 3H). ^13^C{^1^H} NMR (151 MHz, CD_2_Cl_2_): δ 130.9, 118.5, 79.3, 79.2, 67.7, 66.9, 40.4, 21.2. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_8_H_12_O_3_Na, 179.0678; found, 179.0676. Specific rotation: [α]D^20^ −11.2 (c 1.00, CH_2_Cl_2_).
Compound 22
To a stirred mixture of 20 (722 mg, 5.2 mmol, 1 equiv) and molecular sieve (4 Å) in dry DCM (40 mL) were added p-toluenesulfonic acid (89 mg, 515 μmol, 0.1 equiv) and anisaldehyde-dimethylacetal (1.22 g, 1.14 mL, 6.7 mmol, 1.3 equiv) at 0 °C. The resulting purple suspension was then stirred for 3 h at room temperature. Once TLC had indicated full completion, the reaction was quenched with sat. NaHCO_3_ solution. The whole mixture was then filtered over Celite, before the product was extracted several times with DCM. The combined organic phases were dried over Na_2_SO_4_, filtered, and concentrated. The obtained crude product was then dissolved in dry DCM (40 mL) and cooled to −40 °C. Then, DIBAL-H (1 M in hexane, 7 mL, 7 mmol, 1.36 equiv) was added dropwise via a syringe at the respective temperature. The resulting mixture was stirred at −40 °C for 1 h, before the reaction was quenched with sat. aqueous NH_4_Cl solution. DCM was added, and the resulting mixture was stirred for 30 min at room temperature (reaction mixture thickens). The organic layer was filtered over a short plug of Celite to remove the solids and concentrated. The crude product was purified via column chromatography (petroleum ether/ethyl acetate, 6:1) to give 1.11 g (83% over 2 steps) of the PMB-protected alcohol 22 as colorless oil. ^1^H NMR (400 MHz, CDCl_3_): δ 7.23–7.15 (m, 2H), 6.84–6.76 (m, 2H), 5.79 (ddt, J = 17.3, 10.2, 7.2 Hz, 1H), 5.16–5.03 (m, 2H), 4.46–4.36 (m, 2H), 4.31 (dd, J = 4.6, 2.3 Hz, 1H), 3.72 (s, 3H), 2.57 (m, 1H), 2.46 (m, 2H), 2.40 (d, J = 2.3 Hz, 1H), 1.32 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 159.3, 132.9, 130.7, 129.3, 118.8, 114.0, 82.0, 79.5, 74.5, 67.4, 64.5, 55.4, 39.4, 18.3. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_16_H_20_O_3_Na, 283.1304; found, 283.1307. Specific rotation: [α]D^20^ +46.7 (c 1.00, CH_2_Cl_2_).
Compound 25 (Preparation Out of 22)
The starting material 22 (65 mg, 250 μmol, 1 equiv) was dissolved in dry toluene (25 mL) before the reaction mixture was degassed via freeze–pump–thaw cycles (3×). After addition of the Grubbs second generation catalyst (11 mg, 12 μmol, 0.05 equiv), an ethylene atmosphere was created, which was maintained throughout the reaction. The slightly pink homogeneous solution was stirred for 2 h at 55 °C (oil bath). As soon as TLC had indicated complete conversion, the reaction was quenched by adding basic l-cysteine solution (5 equiv. cysteine in 20 mL 1 M NaOH) and stirred for 16 h at room temperature. The dark biphasic mixture was separated, and the amber org. phase was washed two times with 1 N NaOH solution, dried over Na_2_SO_4_, filtered over a short plug of silica, and concentrated to obtain crude brown oil, which was purified via column chromatography (petroleum ether/ethyl acetate, 7:1) to give 31 mg (47%) of the product 25 as yellowish oil. ^1^H NMR (400 MHz, CDCl_3_): δ 7.28–7.20 (m, 2H), 6.90–6.82 (m, 2H), 6.41 (ddt, J = 17.8, 11.0, 0.7 Hz, 1H), 5.85–5.78 (m, 1H), 5.50 (ddq, J = 17.8, 1.8, 1.0 Hz, 1H), 5.16 (dq, J = 11.0, 1.3 Hz, 1H), 4.86 (d, J = 6.0 Hz, 1H), 4.54–4.36 (m, 2H), 3.79 (s, 3H), 2.66 (dd, J = 18.0, 2.8 Hz, 1H), 2.50–2.44 (m, 1H), 1.57 (d, J = 6.7 Hz, 1H), 1.45 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 159.1, 142.7, 131.7, 131.5, 131.0, 128.9, 115.6, 113.9, 86.8, 81.5, 65.2, 55.4, 43.5, 19.4. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_16_H_20_O_3_Na, 283.1304; found, 283.1301. Specific rotation: [α]D^20^ −76.0 (c 1.00, CH_2_Cl_2_).
Compound 23
The starting material 22 (770 mg, 2.9 mmol, 1 equiv) was dissolved in dry DCM (30 mL), then pyridine (702 mg, 715 μL, 8.9 mmol, 3 equiv) was added in one portion, and the mixture was chilled to 0 °C. Subsequently, acetyl chloride (580 mg, 530 μL, 7.4 mmol, 2.5 equiv) was added dropwise via a syringe while a white precipitant was formed. After 30 min, H_2_O and sat. NaHCO_3_ solution were added and the product was extracted three times with ethyl acetate. The combined organic phases were washed twice with water and once with brine, dried over Na_2_SO_4_, filtered, and concentrated to obtain 882 mg (99%) of 23 as crude colorless oil, which was used for the next step without further purification. ^1^H NMR (400 MHz, CDCl_3_): δ 7.25–7.21 (m, 2H), 6.89–6.83 (m, 2H), 5.96–5.81 (m, 1H), 5.55 (d, J = 2.3 Hz, 1H), 5.21–5.10 (m, 2H), 4.54–4.43 (m, 2H), 3.79 (s, 3H), 2.54 (m, 2H), 2.49 (d, J = 2.3 Hz, 1H), 2.11 (s, 3H), 1.37 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 169.7, 159.0, 132.6, 131.0, 128.7, 118.7, 113.7, 79.2, 78.2, 75.0, 67.9, 64.4, 55.3, 39.6, 21.0, 19.6. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_18_H_22_O_4_Na, 325.1410; found, 325.1410. Specific rotation: [α]D^20^ +23.5 (c 1.00, CH_2_Cl_2_).
Compound 24
The starting material 23 (882 mg, 2.9 mmol, 1 equiv) was dissolved in dry ethyl acetate (300 mL) before the reaction mixture was degassed via freeze–pump–thaw cycles (3×). After addition of the Grubbs second generation catalyst (124 mg, 146 μmol, 0.05 equiv), an ethylene atmosphere was created, which was maintained throughout the reaction. The slightly pink homogeneous solution was stirred for 2 h at 55 °C (oil bath). As soon as TLC had indicated complete conversion, the reaction mixture was exposed to air to oxidize the remaining catalyst. The reaction mixture was then filtered over a short plug of silica, washed out with ether, before the solvents were distilled off. Crude brown oil was obtained, which was purified via column chromatography (petroleum ether/ethyl acetate 20:1) to yield 827 mg (94%) of the cyclopentane 24 as colorless oil. ^1^H NMR (400 MHz, CDCl_3_): δ 7.28–7.20 (m, 2H), 6.89–6.81 (m, 2H), 6.40 (ddt, J = 17.7, 11.0, 0.7 Hz, 1H), 6.14 (d, J = 1.3 Hz, 1H), 5.99 (t, J = 2.8 Hz, 1H), 5.16–5.05 (m, 2H), 4.55 (d, J = 11.2 Hz, 1H), 4.45 (d, J = 11.2 Hz, 1H), 3.78 (s, 3H), 2.71 (dd, J = 18.6, 3.0 Hz, 1H), 2.56 (ddt, J = 18.6, 2.3, 1.2 Hz, 1H), 2.12 (s, 3H), 1.37 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 170.7, 158.9, 139.7, 134.1, 131.3, 131.1, 128.8, 115.1, 113.8, 85.3, 79.8, 65.1, 55.3, 45.4, 21.1, 19.5. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_18_H_22_O_4_Na, 325.1410; found, 325.1414. Specific rotation: [α]D^20^ −31.4 (c 1.00, CH_2_Cl_2_).
Compound 25 (Preparation Out of 24)
To a solution of the ester 24 (874 mg, 2.9 mmol, 1 equiv) in MeOH (30 mL) was added potassium carbonate (800 mg, 5.8 mmol, 2 equiv) in one portion. The resulting white suspension was stirred at room temperature for 16 h. The solvent was completely removed under reduced pressure, and the residue was purified via flash column chromatography (petroleum ether/ethyl acetate, 7:1) to yield 750 mg (quant.) of the cyclic allylic alcohol 25 as pure white crystals. ^1^H NMR (400 MHz, CDCl_3_): δ 7.28–7.20 (m, 2H), 6.90–6.82 (m, 2H), 6.41 (ddt, J = 17.8, 11.0, 0.7 Hz, 1H), 5.85–5.78 (m, 1H), 5.50 (ddq, J = 17.8, 1.8, 1.0 Hz, 1H), 5.16 (dq, J = 11.0, 1.3 Hz, 1H), 4.86 (d, J = 6.0 Hz, 1H), 4.54–4.36 (m, 2H), 3.79 (s, 3H), 2.66 (dd, J = 18.0, 2.8 Hz, 1H), 2.50–2.44 (m, 1H), 1.57 (d, J = 6.7 Hz, 1H), 1.45 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 159.1, 142.7, 131.7, 131.5, 131.0, 128.9, 115.6, 113.9, 86.8, 81.5, 65.2, 55.4, 43.5, 19.4. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_16_H_20_O_3_Na, 283.1304; found, 283.1301. Specific rotation: [α]D^20^ −76.0 (c 1.00, CH_2_Cl_2_). Melting point: mp 59.1–61.2 °C.
Compound 26
To a solution of 25 (300 mg, 1.15 mmol, 1 equiv) in dry DCM (5 mL) at 0 °C was added VO(acac)2 (61 mg, 230 μmol, 0.2 equiv) in one portion, followed by the dropwise addition of tert-butylhydroperoxide (5.5 M in decane, 230 μL, 1.3 mmol, 1.1 equiv). The resulting red solution was allowed to reach room temperature. After being stirred for 1 h, TLC had indicated complete conversion. The reaction was quenched by the addition of a saturated aqueous solution of Na_2_S_2_O_3_ and a saturated aqueous solution of NH_4_Cl. The aqueous layer was extracted with DCM, the combined organic layers were dried over Na_2_SO_4_, filtered, and reduced in vacuo. The residue was purified by column chromatography (petroleum ether/ethyl acetate, 7:1) to give 222 mg (70%) of the epoxide 26 as colorless oil. ^1^H NMR (600 MHz, CD_2_Cl_2_): δ 7.29–7.22 (m, 2H), 6.91–6.85 (m, 2H), 5.83 (dd, J = 17.5, 10.9 Hz, 1H), 5.58 (dd, J = 17.5, 1.4 Hz, 1H), 5.38 (dd, J = 10.9, 1.4 Hz, 1H), 4.46 (d, J = 8.7 Hz, 1H), 4.44 (d, J = 10.8 Hz, 1H), 4.35 (d, J = 10.7 Hz, 1H), 3.79 (s, 3H), 3.44 (dd, J = 1.9, 0.6 Hz, 1H), 2.26 (d, J = 9.1 Hz, 1H), 2.20 (d, J = 14.6 Hz, 1H), 2.11 (dd, J = 14.4, 2.0 Hz, 1H), 1.36 (d, J = 0.7 Hz, 3H). ^13^C{^1^H} NMR (151 MHz, CD_2_Cl_2_): δ 159.2, 132.8, 131.3, 129.1, 119.0, 113.7, 82.8, 79.8, 67.1, 64.7, 62.6, 55.3, 39.7, 21.3. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_16_H_20_O_4_Na, 299.1254; found, 299.1253. Specific rotation: [α]D^20^ −41.8 (c 1.00, CH_2_Cl_2_).
Compound 27
To a stirred solution of the diol 20 (2.5 g, 17.8 mmol, 1 equiv) in dry DCM (150 mL) was added imidazole (3.0 g, 44.6 mmol, 2.5 equiv) and the resulting mixture was chilled to 0 °C. Subsequently, chlorotriethylsilane (3.0 g, 3.3 mL, 19.6 mmol, 1.1 equiv) was added at the respective temperature, causing the formation of a white precipitant. Stirring was continued for 15 min until TLC had indicated full conversion. Then, the reaction was quenched by the addition of water and the aqueous phase was extracted twice with DCM. The combined organic layers were washed once with brine, dried over Na_2_SO_4_, and concentrated.
The obtained oily crude mixture was redissolved in dry DCM (150 mL), before 2,6-lutidine (4.8 g, 5.2 mL, 44.6 mmol, 2.5 equiv) and tert-butyldimethylsilyl trifluoromethanesulfonate (7.1 g, 6.2 mL, 26.8 mmol, 1.5 equiv) were added at room temperature. The slightly purple solution was stirred for 16 h until TLC had indicated complete conversion. The reaction was then quenched with sat. NH_4_Cl solution, and the aqueous phase was extracted twice with DCM. The combined organic layers were dried over Na_2_SO_4_, filtered, and concentrated. The residue was purified via column chromatography (petroleum ether) to give 5.14 g (78%) of the bis-silylated material 27 as colorless oil. ^1^H NMR (400 MHz, CDCl_3_): δ 5.98–5.81 (m, 1H), 5.10–5.01 (m, 2H), 4.15 (d, J = 2.2 Hz, 1H), 2.41 (m, 2H), 2.34 (d, J = 2.2 Hz, 1H), 1.22 (s, 3H), 0.98 (t, J = 7.9 Hz, 9H), 0.87 (s, 9H), 0.77–0.57 (m, 6H), 0.09 (s, 6H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 134.9, 117.4, 84.1, 77.8, 73.7, 70.5, 42.9, 26.1, 23.7, 18.5, 7.0, 5.0, −1.8, −1.9. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_20_H_40_O_2_Si_2_Na, 391.2459; found, 391.2453. Specific rotation: [α]D^20^ +7.6 (c 1.00, CH_2_Cl_2_).
Compound 28
The starting material 27 (3 g, 8.1 mmol, 1 equiv) was dissolved in dry ethyl acetate (800 mL) before the reaction mixture was degassed via freeze–pump–thaw cycles (3×). After addition of the Grubbs second generation catalyst (345 mg, 407 μmol, 0.05 equiv), an ethylene atmosphere was created, which was maintained throughout the reaction. The slightly pink homogeneous solution was stirred for 2 h at 55 °C (oil bath) causing a color change to dark brown. As soon as TLC had indicated complete conversion, the reaction mixture was exposed to air, to oxidize the remaining catalyst, before the solvent was distilled off. Crude brown oil was obtained, which was purified via column chromatography (petroleum ether) to yield 2.49 g (83%) of the cyclopentane 28. ^1^H NMR (400 MHz, CDCl_3_): δ 6.36–6.23 (m, 1H), 5.76 (dq, J = 3.0, 2.1, 1.6 Hz, 1H), 5.40 (ddq, J = 17.8, 1.7, 0.8 Hz, 1H), 5.07 (ddq, J = 11.0, 1.5, 0.8 Hz, 1H), 4.62 (p, J = 1.3 Hz, 1H), 2.46–2.33 (m, 2H), 1.28 (s, 3H), 1.04–0.90 (m, 9H), 0.85 (s, 9H), 0.76–0.62 (m, 6H), 0.08 (s, 3H), 0.07 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 143.3, 132.0, 127.8, 114.6, 85.2, 77.4, 45.8, 26.0, 23.7, 18.1, 7.2, 5.5, −2.2, −2.5. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_20_H_40_O_2_Si_2_Na, 391.2459; found, 391.2456. Specific rotation: [α]D^20^ −47.4 (c 1.00, CH_2_Cl_2_).
Compound 29
To a solution of 28 (2.5 g, 6.8 mmol, 1 equiv) in THF (60 mL) and H_2_O (10 mL) was added p-toluenesulfonic acid (117 mg, 678 μmol, 0.1 equiv). The mixture was then stirred at room temperature until TLC had indicated full conversion (5 h). Subsequently, saturated aqueous NaHCO_3_ solution was added and the aqueous phase was extracted with ether. The combined organic layers were washed with H_2_O and brine, dried over Na_2_SO_4_, filtered, and concentrated. The residue was purified via column chromatography (petroleum ether/ethyl acetate, 12:1) to give 1.63 g (94%) of the allylic alcohol 29 as white crystals. ^1^H NMR (400 MHz, CDCl_3_): δ 6.41 (ddt, J = 17.7, 10.9, 0.7 Hz, 1H), 5.79 (t, J = 2.8 Hz, 1H), 5.45 (ddq, J = 17.8, 1.7, 0.9 Hz, 1H), 5.17–5.12 (m, 1H), 4.53 (d, J = 6.6 Hz, 1H), 2.55–2.36 (m, 2H), 1.42 (d, J = 6.6 Hz, 1H), 1.39 (s, 3H), 0.83 (s, 9H), 0.08 (s, 3H), 0.06 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 145.3, 132.0, 131.6, 115.2, 83.7, 77.2, 46.5, 25.8, 23.4, 18.1, −2.4, −2.5. HRMS (ESI) m/z: [M – H]^−^ calcd for C_14_H_25_O_2_Si, 253.1629; found, 253.1633. Specific rotation: [α]D^20^ −57.5 (c 1.00, CH_2_Cl_2_). Melting point: mp 50.5–52.3 °C.
Compound 30
To a solution of 29 (1.28 g, 5.0 mmol, 1 equiv) in dry DCM (50 mL) at 0 °C was added VO(acac)2 (267 mg, 1.0 mmol, 0.2 equiv) in one portion, followed by the dropwise addition of tert-butylhydroperoxide (5.5 M in decane, 1 mL, 5.5 mmol, 1.1 equiv). The resulting red solution was allowed to reach room temperature. After being stirred for 1 h, TLC had indicated complete conversion. The reaction was quenched by the addition of a saturated aqueous solution of Na_2_S_2_O_3_ and a saturated aqueous solution of NH_4_Cl. The aqueous layer was extracted with DCM, the combined organic layers were dried over Na_2_SO_4_, filtered, and reduced in vacuo. The residue was purified by column chromatography (petroleum ether/ethyl acetate, 10:1) to give 1.06 g (78%) of the epoxide 30 as white crystals. ^1^H NMR (400 MHz, CDCl_3_): δ 5.82 (dd, J = 17.5, 10.8 Hz, 1H), 5.57 (dd, J = 17.5, 1.3 Hz, 1H), 5.37 (dd, J = 10.8, 1.3 Hz, 1H), 4.17 (dd, J = 9.8, 0.7 Hz, 1H), 3.45 (dd, J = 2.2, 0.7 Hz, 1H), 2.08 (dd, J = 14.7, 0.7 Hz, 1H), 2.00 (dd, J = 14.7, 2.2 Hz, 1H), 1.85 (d, J = 9.9 Hz, 1H), 1.28 (s, 3H), 0.85 (s, 9H), 0.10 (s, 3H), 0.09 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 132.9, 119.2, 82.4, 81.3, 68.2, 64.0, 42.5, 25.8, 25.4, 18.0, −2.2, −2.3. HRMS (ESI) m/z: [M
- Na]^+^ calcd for C_14_H_26_O_3_SiNa, 293.1543; found, 293.1539. Specific rotation: [α]D^20^ −16.2 (c 1.00, CH_2_Cl_2_). Melting point: mp 52.1–53.8 °C.
Compound 31
To a stirred solution of the allylic alcohol 30 (1.06 g, 3.9 mmol, 1 equiv) in dry DMF (4 mL) were added imidazole (640 mg, 9.4 mmol, 2.4 equiv) and tert-butyldimethylsilyl chloride (709 mg, 4.7 mmol, 1.2 equiv) at room temperature. Stirring was continued for 15 h until TLC had indicated full conversion. Then, the reaction was quenched by the addition of water. Subsequently, diethyl ether (100 mL) was added and the organic phase was extracted five times with water (5 mL) to remove the DMF. The ether phase was then dried over Na_2_SO_4_, filtered, and concentrated. The residue was purified via column chromatography (petroleum ether/toluene, 2:1) to give 1.5 g (quant.) of the epoxide 31 as colorless oil. ^1^H NMR (400 MHz, CDCl_3_): δ 5.96 (dd, J = 17.2, 10.8 Hz, 1H), 5.37 (dd, J = 17.2, 1.7 Hz, 1H), 5.23 (dd, J = 10.8, 1.7 Hz, 1H), 4.25 (s, 1H), 3.24–3.19 (m, 1H), 2.19 (d, J = 14.3 Hz, 1H), 1.93 (ddd, J = 14.3, 1.9, 0.9 Hz, 1H), 1.27 (d, J = 0.8 Hz, 3H), 0.90 (s, 9H), 0.86 (s, 9H), 0.11 (s, 3H), 0.10 (s, 3H), 0.09 (s, 3H), 0.07 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 133.0, 117.3, 84.2, 80.2, 66.1, 61.3, 42.2, 27.0, 26.1, 26.0, 18.4, 18.0, −1.9, −2.4, −3.8, −4.5. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_20_H_40_O_3_Si_2_Na, 407.2408; found, 407.2404. Specific rotation: [α]D^20^ −54.8 (c 1.00, CH_2_Cl_2_).
Compound 32
A Schlenk flask containing Pd_2_(dba)3 (200 mg, 218 μmol, 0.06 equiv) and (R,R)-DACH ligand [138517-61-0] (377 mg, 546 μmol, 0.15 equiv) was charged with dry degassed DCM (freeze–pump–thaw) (30 mL). After 5 min, the color of the solution had changed from dark purple to slightly yellow. Then, triethylamine (1.9 g, 2.7 mL, 19.1 mmol, 5.25 equiv) was added at 0 °C, directly followed by formic acid (840 mg, 690 μL, 18.2 mmol, 5 equiv). The mixture was allowed to reach room temperature (10 min), before the epoxide 31 (1.4 g, 3.6 mmol, 1 equiv) dissolved in 5 mL of dry degassed DCM was added. A color change to green was observed, and the reaction was stirred until TLC analysis showed total consumption of the starting material (3 h). A sat. aqueous solution of NH_4_Cl was added, and the mixture was extracted with DCM; the organic extracts were dried over Na_2_SO_4_, filtered, and reduced in vacuo. The crude product was purified by column chromatography (toluene), delivering the desired product 32 (1.16 g, 82%) as colorless oil. ^1^H NMR (400 MHz, CDCl_3_): δ 6.16–6.02 (m, 1H), 5.25–5.15 (m, 2H), 4.11 (ddddd, J = 10.7, 6.8, 5.6, 2.3, 1.2 Hz, 1H), 3.67 (dt, J = 3.4, 1.0 Hz, 1H), 2.90 (ddd, J = 9.2, 5.6, 3.4 Hz, 1H), 2.57 (d, J = 11.0 Hz, 1H), 2.31 (ddd, J = 15.0, 6.9, 0.8 Hz, 1H), 1.87 (dd, J = 15.1, 2.3 Hz, 1H), 1.37 (s, 3H), 0.91 (s, 9H), 0.84 (s, 9H), 0.09 (s, 3H), 0.09 (s, 3H), 0.08 (s, 3H), 0.08 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 135.4, 117.7, 86.1, 83.8, 76.3, 52.5, 51.9, 26.1, 25.8, 24.8, 18.1, 18.0, −2.0, −2.3, −3.8, −4.0. HRMS (ESI) m/z: [M + H]^+^ calcd for C_20_H_43_O_3_Si_2_, 387.2745; found, 387.2745. Specific rotation: [α]D^20^ −13.1 (c 1.00, CH_2_Cl_2_).
Compound 33
A solution of the homoallylic alcohol 32 (1.78 g, 4.6 mmol, 1 equiv) in DCM/MeOH (40 mL each) was cooled to −80 °C. Then, a stream of ozone was bubbled through the solution until it took on a deep blue color. After 5 min of further stirring, a stream of oxygen was bubbled through the solution until the blue color had disappeared. Subsequently, triphenylphosphine (1.81 g, 6.9 mmol, 1.5 equiv) was added at −80 °C before the reaction was allowed to reach room temperature. After 30 min of stirring at respective temperature, the solvents were removed in vacuo to give a slightly yellow crude oil containing 33. The obtained crude material was directly used for the next step without further purification. However, an analytical sample was purified via column chromatography (DCM/ether, 15:1) to collect NMR spectra and physical data. ^1^H NMR (400 MHz, CDCl_3_): δ 10.00 (d, J = 2.1 Hz, 1H), 4.64–4.56 (m, 1H), 4.10 (dd, J = 4.2, 1.0 Hz, 1H), 3.05 (ddd, J = 6.2, 4.1, 2.1 Hz, 1H), 2.93 (d, J = 8.7 Hz, 1H), 2.34 (ddd, J = 14.6, 7.1, 1.0 Hz, 1H), 1.95 (dd, J = 14.6, 3.8 Hz, 1H), 1.38 (s, 3H), 0.88 (s, 9H), 0.82 (s, 9H), 0.12 (s, 3H), 0.09 (s, 3H), 0.09 (s, 6H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 204.9, 83.6, 82.9, 73.3, 59.2, 50.6, 26.0, 25.8, 23.8, 18.0, 18.0, −2.1, −2.4, −3.9, −4.4. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_19_H_40_O_4_Si_2_Na, 411.2357; found, 411.2360. Specific rotation: [α]D^20^ −3.7 (c 1.00, CH_2_Cl_2_). Melting point: mp 83.8–84.5 °C.
Compound 6
The crude β-hydroxy aldehyde 33 (1.79 g, 4.6 mmol, 1 equiv) was dissolved in DCM (50 mL) before imidazole (752 mg, 11.1 mmol, 2.4 equiv) and chlorotriethylsilane (830 mg, 930 μL, 5.5 mmol, 1.2 equiv) were added. The reaction was stirred for 15 min until TLC had indicated full conversion. Then, the reaction was quenched by the addition of water, and the aqueous phase was extracted twice with DCM. The combined organic layers were washed once with brine, dried over Na_2_SO_4_, and concentrated. The residue was purified via column chromatography (petroleum ether/DCM, 4:1) to yield 2.08 g (89% over 2 steps) of the desired product 6 as colorless oil. ^1^H NMR (400 MHz, CDCl_3_): δ 9.81 (d, J = 5.2 Hz, 1H), 4.61 (dt, J = 8.5, 7.3 Hz, 1H), 3.91 (d, J = 6.2 Hz, 1H), 2.94 (ddd, J = 8.4, 6.2, 5.2 Hz, 1H), 2.11 (dd, J = 7.3, 0.8 Hz, 2H), 1.34 (s, 3H), 0.91 (t, J = 7.9 Hz, 9H), 0.88 (s, 9H), 0.83 (s, 9H), 0.57–0.49 (m, 6H), 0.10 (s, 3H), 0.08 (s, 3H), 0.05 (s, 3H), −0.01 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 205.8, 83.9, 82.7, 73.4, 58.2, 48.8, 25.9, 25.8, 23.1, 18.1, 18.1, 6.8, 4.8, −2.0, −2.3, −4.2, −4.6. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_25_H_54_O_4_Si_3_Na, 525.3222; found, 525.3216. Specific rotation: [α]D^20^ −31.3 (c 1.00, CH_2_Cl_2_).
Compound 34
To a stirred solution of 14 (100 mg, 306 μmol, 1 equiv) in THF (3 mL) was added tetrakis(triphenylphosphine)palladium (18 mg, 15 μmol, 0.05 equiv). Then, the mixture was cooled to 0 °C and vinylmagnesium bromide (1 M in THF, 920 μL, 920 μmol, 3 equiv) was added dropwise. The mixture was allowed to reach room temperature over a period of 15 min, which caused the formation of a brown precipitate. After 1 h, 5 mL of ether was added before the reaction was quenched with sat. NH_4_Cl solution. The aqueous phase was then extracted three times with ether; the combined organic phases were washed with water and brine. Drying over Na_2_SO_4_ and subsequent evaporation of the solvent furnished a crude mixture, which was purified via column chromatography (petroleum ether/ethyl acetate, 80:1) to yield 66 mg (95%) of the diene 34. ^1^H NMR (400 MHz, CDCl_3_): δ 6.64–6.50 (m, 1H), 5.87 (dd, J = 10.9, 1.2 Hz, 1H), 5.14–4.95 (m, 2H), 3.70 (t, J = 7.0 Hz, 2H), 2.27 (t, J = 6.9 Hz, 2H), 1.78 (d, J = 1.5 Hz, 3H), 0.89 (s, 9H), 0.04 (s, 6H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 136.6, 133.4, 127.3, 115.1, 62.3, 43.3, 26.1, 18.5, 17.3, −5.2. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_13_H_26_OSiNa, 249.1645; found, 249.1640.
Compound 35
To a stirred solution of 34 (118 mg, 395 μmol, 1 equiv) in THF (3 mL) was added 9 BBN (0.5 M in THF, 3.16 mL, 1.58 mmol, 4 equiv) dropwise at 0 °C. The mixture was allowed to reach room temperature and stirred until TLC confirmed full completion after 4 h. Then, K_2_CO_3_ (10% solution in water, 4 mL, 3.16 mmol, 8 equiv) was added, followed by H_2_O_2_ (30 wt %, 290 μL, 2.77 mmol, 7 equiv). The resulting suspension was stirred for 2 h before the reaction was quenched with solid NH_4_Cl. The mixture was extracted three times with ethyl acetate, washed with brine, dried over Na_2_SO_4_, and concentrated. The residue was purified via column chromatography (petroleum ether/ethyl acetate, 5:1) to give 45 mg (36%) of the primary alcohol 35. ^1^H NMR (400 MHz, CDCl_3_): δ 5.16 (tq, J = 7.4, 1.3 Hz, 1H), 3.68 (t, J = 6.8 Hz, 2H), 3.62 (q, J = 6.2 Hz, 2H), 2.29 (dddd, J = 7.3, 6.4, 5.6, 0.8 Hz, 2H), 2.23 (td, J = 6.8, 1.0 Hz, 2H), 1.66 (dt, J = 1.5, 0.8 Hz, 3H), 1.43 (t, J = 5.8 Hz, 1H), 0.89 (s, 9H), 0.04 (s, 6H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 136.2, 122.0, 62.4, 62.2, 43.2, 31.7, 26.1, 18.5, 16.6, −5.1. HRMS (ESI) m/z: [M
- Na]^+^ calcd for C_13_H_28_O_2_SiNa, 267.1751; found, 267.1754.
Compounds 37 and 38
To a stirred mixture of 14 (85 mg, 260 μmol, 1 equiv) in dry DMF (3 mL), tetraethylammonium chloride (45 mg, 260 μmol, 1 equiv) and bis(triphenylphosphine)palladium dichloride (9.2 mg, 13 μmol, 0.05 equiv) were added, followed by cis-tributyl(2-ethoxyethenyl)stannane (146 mg, 135 μL, 404 μmol, 1.55 equiv). The resulting mixture was heated to 80 °C (oil bath) and stirred for 45 min until TLC had confirmed full completion. The reaction was quenched by the addition of aqueous NH_4_Cl solution and filtered over a short plug of Celite. The filtrate was poured into a separatory funnel, and the aqueous phase was extracted with ethyl acetate three times. The combined organic phases were washed with water and brine, dried over Na_2_SO_4_, and concentrated. The residue was purified via column chromatography (petroleum ether/ethyl acetate, 60:1) to yield 28 mg (40%) of the coupled product 37, accompanied by 18 mg (29%) of the side product 38. ^1^H NMR (400 MHz, CD_2_Cl_2_): δ 6.14 (dp, J = 11.3, 1.2 Hz, 1H), 5.94 (ddd, J = 6.3, 1.2, 0.6 Hz, 1H), 5.15 (dd, J = 11.3, 6.4 Hz, 1H), 3.83 (q, J = 7.1 Hz, 2H), 3.68 (t, J = 7.0 Hz, 2H), 2.32–2.22 (m, 2H), 1.70 (d, J = 0.8 Hz, 3H), 1.25 (t, J = 7.1 Hz, 3H), 0.88 (s, 9H), 0.04 (s, 6H). ^13^C{^1^H} NMR (101 MHz, CD_2_Cl_2_): δ 145.0, 132.4, 119.3, 103.5, 68.5, 62.8, 43.6, 26.1, 18.6, 17.0, 15.5, −5.2. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_15_H_30_O_2_SiNa, 293.1907; found, 293.1912.
Side product 38 (ketone): ^1^H NMR (400 MHz, CD_2_Cl_2_): δ 6.10 (q, J = 1.2 Hz, 1H), 3.75 (t, J = 6.4 Hz, 2H), 2.31 (td, J = 6.4, 1.0 Hz, 2H), 2.13 (s, 3H), 2.11 (d, J = 1.3 Hz, 3H), 0.88 (s, 9H), 0.04 (s, 6H). ^13^C{^1^H} NMR (101 MHz, CD_2_Cl_2_): δ 198.7, 155.4, 125.5, 61.5, 44.5, 31.9, 26.0, 19.5, 18.5, −5.3. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_13_H_26_O_2_SiNa, 265.1594; found, 265.1596.
Compound 41
Zinc powder (2 g, 30.76 mmol, 1 equiv) was weighed into a three necked round-bottom flask equipped with a condenser and septum and fused under argon. Next, 20 mL of dry THF was added and the resulting gray suspension was subsequently treated with trimethylsilyl chloride (400 μL, 3.08 mmol, 0.1 equiv). The mixture was then heated to 60 °C (oil bath), before tert-butyl bromoacetate (6 g, 4.54 mL, 30.76 mmol, 1 equiv) was added dropwise. Once the addition was complete, a yellow-greenish suspension with some white precipitate was obtained. To determine the concentration of the organyle in the supernatant solution, a small equivalent was taken via a syringe and titrated against iodine until a color change from purple to colorless was observed.
To a suspension of tetrakis(triphenylphosphine)palladium (460 mg, 400 μmol, 0.04 equiv) and lithium chloride (1.27 g, 30 mmol, 3 equiv) in dry THF (4 mL) was added 14 (3.26 g, 10 mmol, 1 equiv) in dry THF (10 mL). The resulting orange suspension was treated with the prepared solution of the zinc organyle 40 (0.83 M in THF, 36 mL, 30 mmol, 3 equiv), followed by the addition of THF (10 mL) and freshly distilled DMPU (24 mL). It was important that the ratio of THF/DMPU roughly equaled 2.5/1. Then, the mixture was heated to 60 °C (oil bath) and stirred for 45 min until TLC had confirmed full conversion. The reaction was quenched with sat. NH_4_Cl solution and stirred for another 30 min before the whole mixture was filtered over a plug of Celite and washed with ether. The filtrate was transferred into a separatory funnel, and the aqueous phase was extracted with ether. The combined organic phases were washed with water (10×), dried over Na_2_SO_4_, filtered, and concentrated. The resulting residue was purified via column chromatography (petroleum ether/ethyl acetate, 60:1) to obtain 2.55 g (81%) of the ester 41 as colorless oil. ^1^H NMR (400 MHz, CDCl_3_): δ 5.33 (tq, J = 7.0, 1.4 Hz, 1H), 3.67 (t, J = 7.1 Hz, 2H), 2.94 (dd, J = 7.0, 1.2 Hz, 2H), 2.24 (td, J = 7.1, 1.1 Hz, 2H), 1.64 (d, J = 1.3 Hz, 3H), 1.44 (s, 9H), 0.88 (s, 9H), 0.04 (s, 6H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 171.8, 135.9, 118.3, 80.4, 62.5, 43.0, 35.2, 28.2, 26.1, 18.5, 17.0, −5.1. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_17_H_34_O_3_SiNa, 337.2169; found, 337.2173.
Compound 42
Potassiumosmate dihydrate (22 mg, 60 μmol) and (DHQD)2_PHAL (234 mg, 300 μmol) were added to a mixture of powdered K_3_Fe(CN)6 (9.80 g, 30 mmol) and K_2_CO_3 (4.12 g, 30 mmol). The resulting mixture was ground to afford 14.18 g of AD-mix-β with 3× increased osmate concentration.
To a mechanically stirred suspension of AD-mix-β-(3×) (9 g, 1.4 g/mmol) in t-BuOH/H_2_O (10 mL each) was added methanesulfonamide (1.83 g, 19.27 mmol, 3 equiv). After 2 h of stirring, the mixture was cooled to 0 °C before compound 41 (2.02 g, 6.42 mmol, 1 equiv) was added. The orange suspension was then stirred for 4 days until TLC had indicated complete conversion. During this period, the color of the reaction mixture gradually changed from orange to yellow. The reaction was quenched with solid Na_2_SO_3_ and allowed to reach room temperature. Ether was added, and the mixture was stirred for 30 min. The product was extracted five times with ether, and the combined organic phases were dried over Na_2_SO_4_, filtered, and concentrated to obtain a crude product, which was purified via column chromatography (petroleum ether/ethyl acetate, 5:1) to yield 1.63 g (73%) of the diol 42 as colorless oil. ^1^H NMR (400 MHz, CDCl_3_) δ 3.95–3.81 (m, 3H), 3.73 (s, 1H), 3.42 (d, J = 4.0 Hz, 1H), 2.50 (dd, J = 15.6, 3.0 Hz, 1H), 2.35 (dd, J = 15.6, 9.9 Hz, 1H), 1.84 (ddd, J = 14.6, 8.8, 4.5 Hz, 1H), 1.72–1.59 (m, 1H), 1.45 (s, 9H), 1.17 (s, 3H), 0.89 (s, 9H), 0.08 (s, 6H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 172.5, 81.0, 74.0, 73.9, 60.2, 39.2, 37.7, 28.2, 26.0, 22.8, 18.2, −5.4, −5.5. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_17_H_36_O_5_SiNa, 371.2224; found, 371.2223. Specific rotation: [α]D^20^ +12.3 (c 1.00, CH_2_Cl_2_).
Compound 43
To a stirred mixture of the diol 42 (300 mg, 860 μmol, 1 equiv) and molecular sieve (4 Å) in dry DCM were added p-toluenesulfonic acid (15 mg, 86 μmol, 0.1 equiv) and 2,2-dimethoxypropane (269 mg, 320 μL, 2.58 mmol, 3 equiv) at 0 °C. The resulting suspension was stirred for 8 h at the respective temperature. Once TLC had indicated full completion, the reaction was quenched with sat. NaHCO_3_-solution. The whole mixture was filtered over a plug of Celite before the product was extracted several times with DCM. The combined organic phases were dried over Na_2_SO_4_, filtered, and concentrated. The crude product was purified via column chromatography (petroleum ether/ethyl acetate, 12:1) to obtain 310 mg (93%) of the acetal protected product 43 as colorless oil. ^1^H NMR (400 MHz, CDCl_3_): δ 4.28 (dd, J = 7.2, 5.6 Hz, 1H), 3.86–3.69 (m, 2H), 2.46 (s, 1H), 2.44 (d, J = 1.7 Hz, 1H), 1.87–1.71 (m, 2H), 1.46 (s, 9H), 1.41 (s, 3H), 1.35 (s, 3H), 1.09 (s, 3H), 0.88 (s, 9H), 0.05 (s, 6H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 170.2, 107.2, 81.3, 81.0, 78.3, 59.3, 42.1, 36.2, 28.7, 28.3, 26.9, 26.1, 21.9, 18.4, −5.2, −5.2. HRMS (ESI) m/z: [M
- Na]^+^ calcd for C_20_H_40_O_5_SiNa, 411.2537; found, 411.2539. Specific rotation: [α]D^20^ +32.1 (c 1.00, CH_2_Cl_2_).
Compound 44
To a solution of 43 (300 mg, 772 μmol, 1 equiv) in THF (6 mL) and H_2_O (1 mL) was added p-toluenesulfonic acid (13 mg, 77 μmol, 0.1 equiv). The mixture was then stirred at room temperature until TLC had indicated full conversion (24 h). Subsequently, saturated aqueous NaHCO_3_ solution was added and the aqueous phase was extracted with ether. The combined organic layers were washed with H_2_O and brine, dried over Na_2_SO_4_, filtered, and concentrated to give 191 mg (90%) of the primary alcohol 44 as colorless oil. The material was used in the next step without further purification. ^1^H NMR (400 MHz, CDCl_3_): δ 4.27 (ddd, J = 8.1, 5.1, 1.1 Hz, 1H), 3.83 (dqd, J = 17.4, 6.2, 3.1 Hz, 2H), 2.86 (t, J = 5.5 Hz, 1H), 2.55 (ddd, J = 15.6, 8.0, 1.2 Hz, 1H), 2.36 (ddd, J = 15.8, 5.1, 0.8 Hz, 1H), 1.79 (t, J = 5.5 Hz, 2H), 1.45 (d, J = 1.1 Hz, 9H), 1.42 (s, 3H), 1.38 (s, 3H), 1.14 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 170.0, 107.7, 82.7, 81.4, 78.5, 59.3, 40.1, 36.1, 28.6, 28.2, 26.8, 21.6. HRMS (ESI) m/z: [M – H]^−^ calcd for C_14_H_25_O_5_, 273.1707; found, 273.1711. Specific rotation: [α]D^20^ −10.4 (c 1.00, CH_2_Cl_2_).
Compound 45
To a stirred solution of the primary alcohol 44 (190 mg, 693 μmol, 1 equiv) in DCM (8 mL) were added solid NaHCO_3_ (175 mg, 2.1 mmol, 3 equiv) and Dess–Martin periodinane (352 mg, 831 μmol, 1.2 equiv) at room temperature. The reaction mixture slightly warmed up and was stirred until TLC had indicated full conversion (30 min). The suspension was then directly filtered over silica (10 g) and eluted with ether. The product containing fractions were combined, and the solvents were distilled off to give the crude aldehyde 45 as a colorless liquid. The obtained crude material was directly used for the next step without further purification. However, an analytical sample was purified via column chromatography (pentane/ether, 5:1), to collect NMR spectra and physical data. ^1^H NMR (400 MHz, CDCl_3_): δ 9.86 (t, J = 2.7 Hz, 1H), 4.28 (dd, J = 7.8, 5.5 Hz, 1H), 2.61 (s, 1H), 2.60 (s, 1H), 2.58 (dd, J = 15.9, 7.8 Hz, 1H), 2.46 (dd, J = 15.9, 5.5 Hz, 1H), 1.46 (s, 9H), 1.44 (s, 3H), 1.37 (s, 3H), 1.21 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 201.2, 169.8, 108.2, 81.5, 80.1, 78.6, 52.3, 36.2, 28.6, 28.2, 26.8, 22.2. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_14_H_24_O_5_Na, 295.1516; found, 295.1517. Specific rotation: [α]D^20^ +20.8 (c 1.00, CH_2_Cl_2_).
Compound 46
To a stirred suspension of methyltriphenylphosphonium iodide (364 mg, 900 μmol, 1.3 equiv), which was dried by coevaporation with toluene before use, in dry ether (5 mL) at 0 °C was added KOtBu (78 mg, 693 μmol, 1 equiv). The resulting orange suspension was stirred for 45 min at 0 °C, before it was added to a solution of the aldehyde 45 (189 mg, 693 μmol, 1 equiv) in dry ether (2 mL) at −20 °C. After the reaction had been stirred for 30 min at −20 °C, TLC indicated complete conversion. The reaction was quenched with sat. NH_4_Cl solution and extracted twice with Et_2_O. The combined organic layers were washed once with water and brine, dried over Na_2_SO_4_, filtered, and concentrated. The residue was chromatographed on silica gel (pentane/ether, 10:1) to provide 133 mg (71% over 2 steps) of the olefin 46 as a colorless liquid. ^1^H NMR (400 MHz, CDCl_3_): δ 5.86 (ddt, J = 17.0, 10.3, 7.3 Hz, 1H), 5.15–5.00 (m, 2H), 4.23 (dd, J = 8.5, 4.5 Hz, 1H), 2.47 (dd, J = 15.7, 8.5 Hz, 1H), 2.34 (dd, J = 15.7, 4.5 Hz, 1H), 2.31–2.27 (m, 2H), 1.45 (s, 9H), 1.41 (s, 3H), 1.34 (s, 3H), 1.08 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 170.2, 133.5, 118.5, 107.4, 81.5, 81.1, 77.8, 43.8, 36.7, 28.7, 28.2, 27.0, 21.7. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_15_H_26_O_4_Na, 293.1723; found, 293.1723. Specific rotation: [α]D^20^ +48.0 (c 1.00, CH_2_Cl_2_).
Compound 9 (Preparation Out of 46)
To a stirred solution of 46 (110 mg, 407 μmol, 1 equiv) in dry DCM (5 mL) was added DIBAL-H (1 M in hexane, 430 μL, 430 μmol, 1.05 equiv) dropwise at −80 °C. The mixture was stirred for 1 h at respective temperature before the reaction was quenched by the addition of methanol and sat. aqueous Na–K-tartrate solution. The resulting suspension was then allowed to reach room temperature before the product was extracted three times with DCM. The combined organic phases were washed with water, dried over Na_2_SO_4_, filtered, and concentrated (40 °C, 300 mbar). The crude product was purified via column chromatography (pentane/ether, 6:1) to yield 69 mg (86%) of the aldehyde 9 as a colorless, volatile liquid. ^1^H NMR (400 MHz, CDCl_3_): δ 9.81 (t, J = 2.0 Hz, 1H), 5.84 (ddt, J = 16.9, 10.3, 7.3 Hz, 1H), 5.17–5.06 (m, 2H), 4.32 (dd, J = 9.6, 3.3 Hz, 1H), 2.66 (ddd, J = 16.4, 9.6, 2.3 Hz, 1H), 2.46 (ddd, J = 16.4, 3.4, 1.8 Hz, 1H), 2.42–2.25 (m, 2H), 1.43 (s, 3H), 1.36 (s, 3H), 1.11 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 200.0, 133.2, 118.8, 107.9, 81.5, 76.0, 44.2, 43.8, 28.7, 27.0, 21.9. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_11_H_18_O_3_Na, 221.1148; found, 221.1149. Specific rotation: [α]D^20^ +8.6 (c 1.00, CH_2_Cl_2_).
Compound 9 (Preparation Out of 10)
A degassed (freeze–pump–thaw) mixture of H_2_O/acetone (1/25) (20 mL) was added to a Schlenk-flask containing the ruthenium catalyst 47 [776230-17-2] (330 mg, 333 μmol, 0.04 equiv) to obtain an orange solution. The whole was then transferred to a separate Schlenk-flask containing alkyne 10 (1.5 g, 8.3 mmol, 1 equiv), and the resulting mixture was heated to 60 °C (oil bath). After 18 h, ether was added, followed by solid Na_2_SO_4_, and the supernatant solution was filtered over a plug of silica. After removal of all volatiles under reduced pressure (40 °C, 300 mbar), the residue was purified via column chromatography (pentane/ether, 6:1) to obtain 1.31 g (79%) of the aldehyde 9 as a slightly yellow, volatile liquid. ^1^H NMR (400 MHz, CDCl_3_): δ 9.81 (t, J = 2.0 Hz, 1H), 5.84 (ddt, J = 16.9, 10.3, 7.3 Hz, 1H), 5.17–5.06 (m, 2H), 4.32 (dd, J = 9.6, 3.3 Hz, 1H), 2.66 (ddd, J = 16.4, 9.6, 2.3 Hz, 1H), 2.46 (ddd, J = 16.4, 3.4, 1.8 Hz, 1H), 2.42–2.25 (m, 2H), 1.43 (s, 3H), 1.36 (s, 3H), 1.11 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 200.0, 133.2, 118.8, 107.9, 81.5, 76.0, 44.2, 43.8, 28.7, 27.0, 21.9. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_11_H_18_O_3_Na, 221.1148; found, 221.1149. Specific rotation: [α]D^20^ +10.2 (c 1.00, CH_2_Cl_2_).
Compound 48
A 20 mL Schlenk flask was charged with Zn(OTf)2 (810 mg, 2.23 mmol, 3 equiv) and (+)-N-methylephedrine (413 mg, 2.30 mmol, 3.1 equiv). To the flask were added dry toluene (6 mL) and triethylamine (233 mg, 319 μL, 2.30 mmol, 3.1 equiv). The resulting slurry was vigorously stirred for 3 h to obtain a cloudy, biphasic mixture before trimethylsilylacetylene (226 mg, 319 μL, 2.30 mmol, 3.1 equiv) was added in one portion. After 30 min of stirring, a solution of the aldehyde 9 (147 mg, 742 μmol, 1 equiv) in dry toluene (1 mL) was added via a syringe. After stirring for 14 h at room temperature, the reaction was quenched by the addition of saturated aqueous NH_4_Cl solution. The reaction mixture was poured into a separatory funnel containing ether. The layers were separated, and the aqueous layer was extracted with ether three times. The combined organic layers were washed with brine, dried over Na_2_SO_4_, and concentrated in vacuo. The obtained residue was taken up in MeOH (10 mL), before K_2_CO_3_ (21 mg, 148 μmol, 0.2 equiv) was added in one portion. As soon as TLC had indicated complete conversion (2 h), the reaction mixture was concentrated and subjected directly to a column chromatography (petroleum ether/ethyl acetate, 12:1) to afford 121 mg (73%) of the secondary propargylic alcohol 48. ^1^H NMR (400 MHz, CDCl_3_): δ 5.85 (ddt, J = 16.9, 10.3, 7.4 Hz, 1H), 5.17–5.03 (m, 2H), 4.63 (dddd, J = 8.4, 5.8, 3.3, 2.2 Hz, 1H), 4.33 (dd, J = 10.8, 2.1 Hz, 1H), 3.02 (d, J = 8.4 Hz, 1H), 2.49 (d, J = 2.2 Hz, 1H), 2.40–2.24 (m, 2H), 1.96 (ddd, J = 14.2, 10.8, 3.4 Hz, 1H), 1.75 (ddd, J = 14.3, 6.1, 2.1 Hz, 1H), 1.44 (s, 3H), 1.37 (s, 3H), 1.11 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 133.3, 118.7, 107.9, 84.2, 81.8, 78.2, 73.3, 60.8, 43.9, 36.3, 28.7, 27.1, 21.8. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_13_H_20_O_3_Na, 247.1304; found, 247.1300. Specific rotation: [α]D^20^ +7.6 (c 1.00, CH_2_Cl_2_).
Compound 7 (Preparation Out of 48)
Methyl lithium (1.6 M in diethyl ether, 280 μL, 446 μmol, 1 equiv) was added to a solution of propargylic alcohol 48 (100 mg, 446 μmol, 1 equiv) in THF (1.5 mL) at −80 °C. After 20 min, the solution was warmed up to room temperature and was ready for use.
During this time, ZnCl_2_ (365 mg, 2.7 mmol, 6 equiv) was weighed to another flask and fused under vacuum. After the flask cooled to room temperature, Cp_2_ZrHCl (253 mg, 981 μmol, 2.2 equiv) and THF (1.0 mL) were added sequentially. The resulting mixture was stirred until all Cp_2_ZrHCl dissolved (about 5 min). The prepared solution of the alkoxide was then transferred via a syringe into the mixture of ZnCl_2_ and Cp_2_ZrHCl in THF, followed by rinsing with THF (0.5 mL). The resulting clear solution was stirred for 2 h and gave a mixture with some gray precipitate. Anhydrous acetonitrile (0.26 mL, 5.0 mmol) was then added to quench the remaining Cp_2_ZrHCl. After 10 min, the reaction was cooled to −80 °C and a solution of I_2_ (226 mg, 892 μmol, 2 equiv) in 1.5 mL of THF was added dropwise. After 1 h at this temperature, an aqueous solution of Na_2_S_2_O_3_ in saturated aqueous NaHCO_3_ solution was added to quench the excess I_2_. After dilution with ether, the reaction mixture was separated and the aqueous layer was extracted with ether. The combined organic phases were dried over Na_2_SO_4_, concentrated, and purified by column chromatography (petroleum ether/ethyl acetate, 12:1) to afford 80 mg (51%) of the α-vinyl iodide 7. ^1^H NMR (400 MHz, CDCl_3_): δ 6.51 (t, J = 1.6 Hz, 1H), 5.93 (dd, J = 1.7, 1.1 Hz, 1H), 5.91–5.78 (m, 1H), 5.18–5.08 (m, 2H), 4.26 (dddd, J = 7.7, 6.2, 3.3, 1.6 Hz, 1H), 4.02 (dd, J = 10.9, 2.0 Hz, 1H), 3.18 (d, J = 7.7 Hz, 1H), 2.41–2.22 (m, 2H), 1.96 (ddd, J = 14.5, 6.1, 2.0 Hz, 1H), 1.80 (ddd, J = 14.4, 10.9, 3.4 Hz, 1H), 1.43 (s, 3H), 1.32 (s, 3H), 1.10 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 133.2, 125.4, 118.9, 115.1, 107.8, 81.9, 77.3, 76.3, 43.7, 33.9, 28.7, 27.1, 21.8. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_13_H_21_IO_3_Na, 375.0427; found, 375.0423. Specific rotation: [α]D^20^ +9.8 (c 1.00, CH_2_Cl_2_).
Compound 49
To a solution of 7 (50 mg, 142 μmol, 1 equiv) in MeOH (2 mL), p-toluenesulfonic acid (5 mg, 28 μmol, 0.2 equiv) was added in one portion. The resulting mixture was heated up to 50 °C (oil bath) and stirred for 24 h at respective temperature. After TLC had indicated complete conversion, the solvent was removed under reduced pressure and the residue was chromatographed on silica gel (petroleum ether/ethyl acetate, 1:1) to yield 35 mg (80%) of the triol 49 as slightly yellow crystals. ^1^H NMR (400 MHz, CDCl_3_): δ 6.53 (t, J = 1.6 Hz, 1H), 5.93 (dd, J = 1.7, 1.0 Hz, 1H), 5.95–5.84 (m, 1H), 5.23–5.13 (m, 2H), 4.32 (tdd, J = 6.7, 3.2, 1.5 Hz, 1H), 3.73 (ddd, J = 10.9, 3.6, 2.0 Hz, 1H), 3.53 (d, J = 6.7 Hz, 1H), 2.84 (dd, J = 3.8, 1.2 Hz, 1H), 2.27 (dt, J = 7.5, 1.1 Hz, 2H), 2.01 (s, 1H), 1.98 (ddd, J = 14.3, 6.7, 2.0 Hz, 1H), 1.74 (ddd, J = 14.3, 10.9, 3.6 Hz, 1H), 1.14 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 133.1, 125.4, 119.9, 115.5, 76.2, 74.2, 73.5, 43.6, 35.2, 21.4. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_10_H_17_IO_3_Na, 335.0114; found, 335.0116. Specific rotation: [α]D^20^ +22.8 (c 1.00, CH_2_Cl_2_). Melting point: mp 97.0–98.2 °C.
Compound 50
To a stirred solution of 7 (40 mg, 114 μmol, 1 equiv) in DCM (2 mL), imidazole (31 mg, 454 μmol, 4 equiv), and chloro tert-butyldimethylsilane (34 mg, 227 μmol, 2 equiv) were added. The reaction was stirred for 48 h until TLC had indicated complete conversion. The mixture was then quenched by the addition of water. The aqueous phase was extracted thrice with DCM, and the combined organic phases were washed with brine, dried over Na_2_SO_4_, and concentrated. The residue was purified via flash chromatography (petroleum ether/ethyl acetate, 25:1) to yield 48 mg (91%) of the TBS-protected alcohol 50 as yellowish oil. ^1^H NMR (400 MHz, CDCl_3_): δ 6.38 (dd, J = 1.5, 0.9 Hz, 1H), 5.91–5.80 (m, 1H), 5.82 (d, J = 1.5 Hz, 1H), 5.15–5.04 (m, 2H), 4.05–3.97 (m, 1H), 3.91–3.83 (m, 1H), 2.32 (ddt, J = 14.1, 7.1, 1.3 Hz, 1H), 2.23 (ddt, J = 14.1, 7.6, 1.2 Hz, 1H), 1.65–1.49 (m, 2H), 1.43 (s, 3H), 1.32 (s, 3H), 1.06 (s, 3H), 0.92 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 133.6, 124.7, 120.2, 118.5, 107.1, 81.6, 76.8, 75.5, 43.6, 38.7, 28.9, 27.2, 26.0, 22.0, 18.3, −4.2, −4.8. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_19_H_35_IO_3_SiNa, 489.1292; found, 489.1292. Specific rotation: [α]D^20^ +18.3 (c 1.00, CH_2_Cl_2_).
Compound 51
A stirred solution of the aldehyde 9 (1.6 g, 8.1 mmol, 1 equiv) in dry toluene (80 mL) was cooled to −80 °C, before vinylmagnesium bromide (1 M in THF, 9.7 mL, 9.7 mmol, 1.2 equiv) was added dropwise. The resulting orange solution was stirred at −80 °C for 1 h, before the reaction was quenched with sat. aqueous NH_4_Cl solution. The aqueous phase was then extracted twice with ether, and the combined organic layers were dried over Na_2_SO_4_, filtered, and concentrated.
The obtained crude material was redissolved in DCM (80 mL), before solid NaHCO_3_ (2 g, 24.2 mmol, 3 equiv) and Dess–Martin periodinane (5.1 g, 12.1 mmol, 1.5 equiv) were added at room temperature. As soon as TLC had indicated full conversion (30 min), the suspension was directly filtered over silica (50 g) and eluted with DCM. The product containing fractions were combined and DCM was distilled off. The residue was purified via column chromatography (pentane/ether, 6:1) to give 1.43 g (79% over 2 steps) of the ketone 51 as a colorless liquid. ^1^H NMR (400 MHz, CDCl_3_): δ 6.42 (dd, J = 17.6, 10.5 Hz, 1H), 6.27 (dd, J = 17.6, 1.1 Hz, 1H), 5.88 (dd, J = 10.5, 1.1 Hz, 1H), 5.92–5.80 (m, 1H), 5.15–5.06 (m, 2H), 4.35 (dd, J = 8.7, 3.7 Hz, 1H), 2.92 (dd, J = 16.2, 8.7 Hz, 1H), 2.58 (dd, J = 16.2, 3.7 Hz, 1H), 2.33 (ddt, J = 7.3, 2.8, 1.2 Hz, 2H), 1.43 (s, 3H), 1.35 (s, 3H), 1.12 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 197.7, 136.6, 133.4, 129.1, 118.6, 107.4, 81.6, 77.0, 43.8, 40.3, 28.7, 27.0, 21.9. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_13_H_20_O_3_Na, 247.1304; found, 247.1306. Specific rotation: [α]D^20^ +4.2 (c 1.00, CH_2_Cl_2_).
Compound 52
To a stirred and light-protected solution of 51 (1.2 g, 5.4 mmol, 1 equiv) in THF (40 mL) and MeCN (10 mL) were added K_2_CO_3_ (2.2 g, 16.1 mmol, 3 equiv), iodine (1.63 g, 6.4 mmol, 1.2 equiv), and quinuclidine (119 mg, 1.1 mmol, 0.2 equiv) sequentially. The initially purple solution quickly turned yellow after the addition of quinuclidine and was stirred until TLC had indicated full completion (45 min). Then, toluene was added (40 mL) and THF as well as MeCN were distilled off (40 °C, 100 mbar). This process was repeated twice to remove most of the THF and MeCN. Subsequently, the toluene-solution was filtered over silica (50 g) and the product was eluted with toluene/ethyl acetate, 8:1. The solvents were removed in vacuo to afford dark yellow, crude oil. The obtained crude material containing 52 was directly used for the next step without further purification. However, an analytical sample was purified via column chromatography (pentane/ether, 6:1), to collect NMR spectra and physical data. ^1^H NMR (400 MHz, CDCl_3_): δ 7.30 (d, J = 2.6 Hz, 1H), 6.87 (d, J = 2.6 Hz, 1H), 5.86 (ddt, J = 16.4, 11.0, 7.3 Hz, 1H), 5.17–5.07 (m, 2H), 4.35 (dd, J = 8.5, 3.7 Hz, 1H), 3.19 (dd, J = 16.4, 8.5 Hz, 1H), 2.77 (dd, J = 16.4, 3.7 Hz, 1H), 2.34 (ddt, J = 7.3, 3.6, 1.2 Hz, 2H), 1.42 (s, 3H), 1.35 (s, 3H), 1.12 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 192.4, 138.7, 133.3, 118.9, 112.5, 107.6, 81.6, 77.4, 43.8, 37.3, 28.7, 27.0, 22.0. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_13_H_19_IO_3_Na 373.0271; found, 373.0273. Specific rotation: [α]D^20^ +6.9 (c 1.00, CH_2_Cl_2_).
Compound 7 (Preparation Out of 52)
To a stirred solution of crude 52 (1.88 g, 5.4 mmol, 1 equiv) in MeOH (60 mL) was added cerium(III) chloride (4.0 g, 16.1 mmol, 3 equiv). The resulting mixture was cooled to −60 °C and then NaBH_4_ (1.0 g, 26.8 mmol, 5 equiv) was added in one portion. After 30 min, TLC had indicated complete conversion and the reaction was quenched by the addition of a sat. aqueous NH_4_Cl solution. As soon as hydrogen evolution had ceased, the mixture was concentrated to a quarter of its volume, before ether was added. The aqueous phase was extracted thrice with ether, the combined organic layers were dried over Na_2_SO_4_, filtered, and concentrated. The residue was purified via column chromatography (petroleum ether/ethyl acetate, 12:1) to yield 1.25 g (66% over 2 steps) of the α-vinyl iodide 7. ^1^H NMR (400 MHz, CDCl_3_): δ 6.51 (t, J = 1.6 Hz, 1H), 5.93 (dd, J = 1.7, 1.1 Hz, 1H), 5.91–5.78 (m, 1H), 5.18–5.08 (m, 2H), 4.26 (dddd, J = 7.7, 6.2, 3.3, 1.6 Hz, 1H), 4.02 (dd, J = 10.9, 2.0 Hz, 1H), 3.18 (d, J = 7.7 Hz, 1H), 2.41–2.22 (m, 2H), 1.96 (ddd, J = 14.5, 6.1, 2.0 Hz, 1H), 1.80 (ddd, J = 14.4, 10.9, 3.4 Hz, 1H), 1.43 (s, 3H), 1.32 (s, 3H), 1.10 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 133.2, 125.4, 118.9, 115.1, 107.8, 81.9, 77.3, 76.3, 43.7, 33.9, 28.7, 27.1, 21.8. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_13_H_21_IO_3_Na, 375.0427; found, 375.0423. Specific rotation: [α]D^20^ +9.1 (c 1.00, CH_2_Cl_2_).
Compound 53
The aldehyde 6 (60 mg, 119 μmol, 1 equiv) and the vinyl iodide 50 (72 mg, 155 μmol, 1.3 equiv) were dissolved in dry ether (2 mL), and the resulting clear solution was cooled to −80 °C. Then, tert-butyllithium (1.7 M in pentane, 180 μL, 310 μmol, 2.6 equiv) was added dropwise at the respective temperature, causing the solution to turn slightly yellow. After 15 min, the reaction was quenched with sat. aqueous NH_4_Cl solution at −80 °C. The aqueous phase was extracted twice with ether, and the combined organic layers were dried over Na_2_SO_4_, filtered, and concentrated. The residue was purified via column chromatography (petroleum ether/ethyl acetate, 20:1) to afford 30 mg (30%) of the coupled product 53 as colorless oil. ^1^H NMR (600 MHz, CDCl_3_): δ 5.86 (ddt, J = 17.4, 10.3, 7.3 Hz, 1H), 5.18–5.04 (m, 4H), 4.55 (d, J = 9.4 Hz, 1H), 4.47 (dd, J = 10.6, 1.6 Hz, 1H), 4.36 (td, J = 7.3, 4.9 Hz, 1H), 4.04 (dd, J = 10.8, 1.5 Hz, 1H), 3.92 (d, J = 3.4 Hz, 1H), 3.18 (d, J = 1.5 Hz, 1H), 2.65 (ddd, J = 9.4, 7.4, 3.5 Hz, 1H), 2.35–2.28 (m, 1H), 2.28–2.21 (m, 1H), 2.15–2.09 (m, 1H), 1.84 (dd, J = 13.5, 4.9 Hz, 1H), 1.74 (ddd, J = 14.1, 10.6, 1.5 Hz, 1H), 1.65 (ddd, J = 14.0, 10.8, 1.7 Hz, 1H), 1.42 (s, 3H), 1.33 (s, 6H), 1.04 (s, 3H), 0.92 (s, 9H), 0.90 (s, 9H), 0.89 (t, J = 8.2 Hz, 9H), 0.84 (s, 9H), 0.49 (q, J = 8.2 Hz, 6H), 0.17 (s, 3H), 0.14 (s, 3H), 0.09 (s, 3H), 0.08 (s, 6H), 0.08 (s, 3H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 150.9, 133.6, 118.4, 112.7, 107.1, 83.4, 81.8, 81.7, 77.6, 75.5, 73.0, 66.4, 50.5, 50.4, 44.0, 36.9, 28.9, 27.2, 26.4, 26.0, 26.0, 24.1, 21.9, 18.6, 18.2, 18.2, 7.1, 5.2, −2.0, −2.3, −3.6, −3.9, −3.9, −4.9. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_44_H_90_O_7_Si_4_Na, 865.5656; found, 865.5660. Specific rotation: [α]D^20^ −50.7 (c 0.50, CH_2_Cl_2_).
Compound 54
To a solution of 53 (30 mg, 35.6 μmol, 1 equiv) in THF (1.2 mL) and H_2_O (200 μL) was added p-toluenesulfonic acid (1.2 mg, 7.1 μmol, 0.2 equiv). The mixture was then stirred at room temperature until TLC had indicated full conversion (48 h). Subsequently, saturated aqueous NaHCO_3_ solution was added and the aqueous phase was extracted with ether. The combined organic layers were washed with H_2_O and brine, dried over Na_2_SO_4_, filtered, and concentrated.
The obtained residue was dissolved in dry DCM (1 mL) before p-toluenesulfonic acid (6 mg, 35.6 μmol, 1 equiv) and 2,2-dimethoxypropane (37 mg, 45 μL, 357 μmol, 10 equiv) were added at room temperature. The resulting suspension was stirred for 1 h at respective temperature. Once TLC had indicated full completion, the reaction was quenched with sat. NaHCO_3_-solution, before the product was extracted several times with DCM. The combined organic phases were dried over Na_2_SO_4_, filtered, and concentrated. The crude product was purified via column chromatography (petroleum ether/ethyl acetate, 20:1) to obtain 19 mg (67%) of the bisketal 54 as colorless oil. ^1^H NMR (600 MHz, CDCl_3_): δ 5.87 (ddt, J = 17.4, 10.2, 7.3 Hz, 1H), 5.31 (s, 1H), 5.30 (s, 1H), 5.12–5.03 (m, 2H), 4.77 (dt, J = 3.9, 1.9 Hz, 1H), 4.46–4.41 (m, 1H), 4.38 (d, J = 9.7 Hz, 1H), 4.00 (d, J = 4.1 Hz, 1H), 3.99 (dd, J = 11.0, 1.5 Hz, 1H), 2.33 (dt, J = 5.0, 4.0 Hz, 1H), 2.29 (ddt, J = 14.0, 7.0, 1.3 Hz, 1H), 2.20 (ddt, J = 14.0, 7.5, 1.2 Hz, 1H), 2.11 (ddd, J = 14.2, 6.8, 0.9 Hz, 1H), 1.95 (ddd, J = 13.9, 11.0, 1.6 Hz, 1H), 1.83 (dd, J = 14.3, 2.4 Hz, 1H), 1.47 (s, 3H), 1.42 (m, 1H), 1.41 (s, 6H), 1.38 (s, 3H), 1.31 (s, 3H), 1.03 (s, 3H), 0.92 (s, 18H), 0.83 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H), 0.06 (s, 3H), 0.06 (s, 3H), 0.01 (s, 3H), 0.01 (s, 3H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 150.4, 133.9, 118.2, 111.0, 107.0, 98.7, 85.3, 82.0, 81.7, 77.9, 73.1, 71.6, 69.5, 46.9, 43.6, 42.8, 40.0, 30.0, 29.0, 27.3, 26.8, 26.0, 26.0, 25.2, 22.2, 19.3, 18.5, 18.2, 18.1, −2.0, −2.1, −2.4, −2.5, −4.4, −4.9. HRMS (ESI) m/z: [M
- Na]^+^ calcd for C_41_H_80_O_7_Si_3_Na, 791.5104; found, 791.5103. Specific rotation: [α]D^20^ −41.6 (c 0.50, CH_2_Cl_2_).
Compound 55
All reagents were titrated, and both starting materials were azeotropically dried with toluene before use.
To a stirred solution of the vinyl iodide 7 (196 mg, 557 μmol, 1.4 equiv) in dry ether (5 mL) at −10 °C was added methylmagnesium bromide (0.9 M in ether, 660 μL, 596 μmol, 1.5 equiv) dropwise. The resulting clear solution was stirred for 30 min at the respective temperature. It was then cooled to −85 °C, before tert-butyllithium (1.6 M in pentane, 700 μL, 1.1 mmol, 2.8 equiv) was added in one portion. The clear solution turned slightly yellow and was stirred for 10 min at −85 °C. Then, the aldehyde 6 (200 mg, 398 μmol, 1 equiv) dissolved in dry ether (2 mL) was added dropwise, causing a color change to dark yellow. After being stirred for another 30 min at −85 °C, the reaction was quenched by the addition of sat. aqueous NH_4_Cl solution and was allowed to reach room temperature. The aqueous phase was extracted thrice with ether, the combined organic layers were dried over Na_2_SO_4_, filtered, and concentrated. The residue was purified via column chromatography (petroleum ether/ethyl acetate, 12:1) to furnish 155 mg (53%) of the coupling product 55, accompanied by 45 mg of the starting aldehyde 6 and 60 mg of the dehalogenated allylic alcohol 56. ^1^H NMR (600 MHz, CDCl_3_): δ 5.88 (ddt, J = 17.5, 10.3, 7.3 Hz, 1H), 5.23 (s, 1H), 5.16 (s, 1H), 5.11–5.05 (m, 2H), 4.49 (dd, J = 8.3, 4.9 Hz, 1H), 4.46–4.41 (m, 1H), 4.29 (td, J = 7.2, 5.1 Hz, 1H), 4.14 (dd, J = 10.4, 2.0 Hz, 1H), 3.93 (d, J = 3.8 Hz, 1H), 3.31 (d, J = 6.8 Hz, 1H), 2.72 (ddd, J = 8.2, 7.1, 3.9 Hz, 1H), 2.64 (d, J = 5.1 Hz, 1H), 2.35–2.24 (m, 2H), 2.11 (ddd, J = 13.6, 7.2, 0.9 Hz, 1H), 1.87–1.80 (m, 2H), 1.75 (ddd, J = 14.0, 8.4, 2.0 Hz, 1H), 1.41 (s, 3H), 1.34 (s, 3H), 1.32 (s, 3H), 1.08 (s, 3H), 0.92 (t, J = 8.0 Hz, 9H), 0.91 (s, 9H), 0.84 (s, 9H), 0.54 (q, J = 8.0 Hz, 6H), 0.15 (s, 3H), 0.09 (s, 3H), 0.09 (s, 3H), 0.09 (s, 3H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 152.2, 133.8, 118.2, 111.2, 107.1, 83.5, 81.8, 81.7, 78.1, 73.2, 72.6, 69.6, 50.7, 49.9, 43.8, 34.9, 28.8, 27.1, 26.2, 25.9, 24.3, 21.7, 18.3, 18.1, 7.1, 4.9, −2.0, −2.4, −3.6, −3.9. HRMS (ESI) m/z: [M - H]^−^ calcd for C_38_H_75_O_7_Si_3_, 727.4826; found, 727.4828. Specific rotation: [α]D^20^ −81.5 (c 0.50, CH_2_Cl_2_) 56: ^1^H NMR (400 MHz, CDCl_3_): δ 5.98–5.77 (m, 2H), 5.31 (dt, J = 17.2, 1.6 Hz, 1H), 5.15 (dt, J = 10.5, 1.5 Hz, 1H), 5.12–5.04 (m, 2H), 4.39 (dtq, J = 9.8, 4.9, 1.6 Hz, 1H), 4.08 (dd, J = 10.7, 2.1 Hz, 1H), 2.39–2.17 (m, 3H), 1.80 (ddd, J = 14.2, 10.7, 3.4 Hz, 1H), 1.55 (ddd, J = 14.2, 7.4, 2.1 Hz, 1H), 1.46–1.41 (m, 3H), 1.39–1.31 (m, 3H), 1.09 (s, 3H). ^13^C{^1^H} NMR (101 MHz, CDCl_3_): δ 140.8, 133.6, 118.4, 114.5, 107.4, 81.8, 77.8, 70.4, 43.8, 36.0, 28.8, 27.1, 21.7. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_13_H_22_O_3_Na 249.1461; found, 249.1464. Specific rotation: [α]D^20^ +7.0 (c 1.00, CH_2_Cl_2_).
Compound 57
The 1,3 diol 55 (250 mg, 343 μmol, 1 equiv) was dissolved in DCM (7 mL) before imidazole (233 mg, 3.4 mmol, 10 equiv) and dichlorodiisopropylsilane (190 mg, 185 μL, 1.0 mmol, 3 equiv) were added. A white precipitate formed, and the reaction mixture was stirred for 45 min until TLC had indicated full conversion. Then, the reaction was quenched by the addition of water and the aqueous phase was extracted twice with DCM. The combined organic layers were dried over Na_2_SO_4_ and concentrated.
The residue was dissolved in THF (6 mL) and H_2_O (1 mL), before p-toluenesulfonic acid (88 mg, 513 μmol, 1.5 equiv) was added. The mixture was then stirred at room temperature until TLC had indicated full conversion (48 h). Subsequently, saturated aqueous NaHCO_3_ solution was added and the aqueous phase was extracted with ether. The combined organic layers were washed with H_2_O and brine, dried over Na_2_SO_4_, filtered, and concentrated. The obtained residue was purified via column chromatography (petroleum ether/ethyl acetate, 10:1) to yield 189 mg (76% over 2 steps) of the secondary alcohol 57 as colorless oil. ^1^H NMR (600 MHz, CDCl_3_): δ 5.89 (ddt, J = 17.4, 10.2, 7.3 Hz, 1H), 5.29 (d, J = 0.9 Hz, 1H), 5.11–5.03 (m, 3H), 4.85 (d, J = 11.1 Hz, 1H), 4.66 (dt, J = 10.2, 1.7 Hz, 1H), 4.25 (dd, J = 9.9, 2.0 Hz, 1H), 4.00 (dt, J = 2.4, 1.0 Hz, 1H), 3.83 (dddt, J = 11.7, 7.2, 4.9, 1.3 Hz, 1H), 2.95 (d, J = 11.6 Hz, 1H), 2.49 (ddd, J = 11.0, 4.8, 2.8 Hz, 1H), 2.36 (ddt, J = 14.0, 7.1, 1.2 Hz, 1H), 2.32–2.25 (m, 2H), 1.84 (dd, J = 15.5, 1.3 Hz, 1H), 1.70 (dddd, J = 42.1, 13.3, 10.1, 2.0 Hz, 2H), 1.41 (s, 3H), 1.40 (s, 3H), 1.31 (s, 3H), 1.11 (s, 3H), 1.02 (dd, J = 7.3, 4.2 Hz, 6H), 0.96 (t, J = 7.2 Hz, 6H), 0.94 (s, 9H), 0.90–0.84 (m, 2H), 0.82 (s, 9H), 0.22 (s, 3H), 0.14 (s, 3H), 0.08 (s, 3H), 0.07 (s, 3H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 148.1, 133.8, 118.2, 111.1, 106.6, 83.7, 83.3, 81.4, 77.2, 74.3, 73.7, 66.8, 52.7, 52.1, 43.6, 33.8, 28.8, 26.8, 26.4, 25.9, 25.8, 21.8, 18.3, 18.0, 17.1, 16.9, 16.9, 16.8, 13.6, 13.2, −2.1, −2.4, −3.9, −4.4. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_38_H_74_O_7_Si_3_Na, 749.4634; found, 749.4631. Specific rotation: [α]D^20^ −60.7 (c 0.50, CH_2_Cl_2_).
Compound 4
The starting material 57 (150 mg, 206 μmol, 1 equiv) was dissolved in DCM (5 mL), before solid NaHCO_3_ (87 mg, 1.0 mmol, 5 equiv) and Dess–Martin periodinane (175 mg, 412 μmol, 2 equiv) were added at room temperature. As soon as TLC had indicated full conversion (30 min), the suspension was directly filtered over silica (10 g) and eluted with DCM. The product containing fractions were combined, and DCM was distilled off. The residue was purified via column chromatography (DCM) to give 139 mg (93%) of the ketone 4 as colorless oil. ^1^H NMR (600 MHz, CDCl_3_): δ 5.88 (ddt, J = 17.3, 10.2, 7.3 Hz, 1H), 5.17 (s, 1H), 5.16 (s, 1H), 5.10–5.03 (m, 2H), 4.62 (d, J = 10.3 Hz, 1H), 4.59 (d, J = 10.1 Hz, 1H), 4.24 (dd, J = 3.6, 1.7 Hz, 1H), 4.21 (dd, J = 10.1, 2.1 Hz, 1H), 3.34 (ddd, J = 10.1, 3.7, 1.1 Hz, 1H), 2.39–2.19 (m, 4H), 1.82 (ddd, J = 13.3, 10.1, 2.1 Hz, 1H), 1.66 (ddd, J = 13.1, 10.6, 2.1 Hz, 1H), 1.45 (s, 3H), 1.38 (s, 3H), 1.30 (s, 3H), 1.11 (s, 3H), 1.06–0.97 (m, 7H), 0.96–0.90 (m, 7H), 0.89 (s, 9H), 0.83 (s, 9H), 0.17 (s, 3H), 0.14 (s, 3H), 0.12 (s, 3H), 0.08 (s, 3H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 212.7, 147.4, 134.0, 118.1, 113.4, 106.6, 81.4, 80.1, 78.5, 77.2, 74.2, 67.8, 58.3, 50.8, 43.7, 34.0, 28.8, 26.8, 26.3, 25.9, 24.2, 21.9, 18.5, 18.0, 17.2, 17.0, 17.0, 16.9, 13.8, 13.4, −2.1, −2.4, −3.8, −4.2. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_38_H_72_O_7_Si_3_Na, 747.4478; found, 747.4481. Specific rotation: [α]D^20^ +40.8 (c 0.50, CH_2_Cl_2_).
Compound 61
To a stirred solution of the ketone 4 (25 mg, 34.5 μmol, 1 equiv) in dry DCM (1 mL) was added trimethylsilyl cyanide (17 mg, 22 μL, 172 μmol, 5 equiv). The resulting clear solution was then chilled to −10 °C, before trimethylsilyl trifluoromethanesulfonate (1 M in DCM, 35 μL, 34.5 μmol, 1 equiv) was added dropwise. The now yellow reaction mixture was allowed to reach room temperature and stirred for 2 h until TLC had indicated complete conversion. Then, sat. aqueous NaHCO_3_ solution was added to quench the reaction and the aqueous phase was extracted twice with DCM. The combined organic layers were dried over Na_2_SO_4_, filtered, and concentrated. The residue was purified via flash column chromatography (petroleum ether/toluene, 1:1) to give 26 mg (91%) of the protected cyanohydrin 61 as colorless oil. ^1^H NMR (600 MHz, CDCl_3_): δ 5.89 (ddt, J = 17.4, 10.2, 7.3 Hz, 1H), 5.48 (s, 1H), 5.15–5.05 (m, 3H), 4.81 (d, J = 10.8 Hz, 1H), 4.74–4.69 (m, 1H), 4.26–4.21 (m, 1H), 3.95 (dd, J = 2.9, 1.6 Hz, 1H), 3.21 (dd, J = 10.9, 2.9 Hz, 1H), 2.61 (d, J = 14.7 Hz, 1H), 2.36 (ddt, J = 14.0, 7.1, 1.2 Hz, 1H), 2.28 (ddt, J = 13.9, 7.6, 1.2 Hz, 1H), 2.14 (dd, J = 14.7, 1.6 Hz, 1H), 1.70–1.62 (m, 2H), 1.37 (s, 3H), 1.34 (s, 3H), 1.30 (s, 3H), 1.09 (s, 3H), 1.02 (dd, J = 7.3, 1.9 Hz, 6H), 0.96 (s, 9H), 0.95–0.92 (m, 7H), 0.88–0.81 (m, 1H), 0.86 (s, 9H), 0.20 (s, 3H), 0.16 (s, 9H), 0.12 (s, 3H), 0.10 (s, 3H), 0.09 (s, 3H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 147.1, 133.9, 122.0, 118.1, 114.5, 106.6, 82.7, 81.5, 81.2, 75.7, 73.7, 77.0, 67.6, 59.6, 56.2, 43.6, 33.8, 28.7, 26.7, 26.3, 26.1, 24.3, 21.8, 18.5, 18.1, 17.2, 17.2, 17.1, 17.0, 14.0, 13.4, 1.3, −2.0, −2.1, −3.9, −4.4. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_42_H_81_NO_7_Si_4_Na, 846.4982; found, 846.4980. Specific rotation: [α]D^20^ +41.6 (c 0.50, CH_2_Cl_2_).
Compound 62
To a stirred solution of the starting material 61 (17 mg, 20.6 μmol, 1 equiv) in dry toluene (800 μL) was added DIBAL-H (1 M in hexane, 103 μL, 103 μmol, 5 equiv) at −80 °C. The resulting clear mixture was then slowly warmed to −50 °C over a period of 2 h. At this point, TLC had indicated full conversion. The reaction was quenched by the addition of water (2 M in THF) and subsequently with sat. aqueous NH_4_Cl solution, before it was allowed to reach room temperature. It was then vigorously stirred for 2 h at room temperature, before the aqueous phase was extracted twice with toluene. The combined organic layers were dried over Na_2_SO_4_, filtered, and concentrated. The residue was purified via column chromatography (petroleum ether/toluene, 2:1) to afford 12 mg (70%) of the aldehyde 62 as colorless oil. ^1^H NMR (600 MHz, CDCl_3_): δ 9.62 (s, 1H), 5.89 (ddt, J = 17.4, 10.2, 7.4 Hz, 1H), 5.10–5.01 (m, 2H), 4.91 (d, J = 1.2 Hz, 1H), 4.83 (s, 1H), 4.80 (d, J = 11.1 Hz, 1H), 4.49 (d, J = 11.4 Hz, 1H), 4.23 (dd, J = 10.4, 1.8 Hz, 1H), 4.07 (dd, J = 3.2, 1.7 Hz, 1H), 3.38 (dd, J = 11.5, 3.2 Hz, 1H), 2.39–2.32 (m, 1H), 2.31–2.24 (m, 1H), 2.21–2.16 (m, 1H), 1.79 (dd, J = 14.9, 1.8 Hz, 1H), 1.72 (ddd, J = 13.2, 10.4, 1.7 Hz, 1H), 1.60–1.53 (m, 1H), 1.39 (s, 3H), 1.35 (s, 3H), 1.31 (s, 3H), 1.09 (s, 3H), 1.03 (dd, J = 7.3, 2.4 Hz, 6H), 0.93 (s, 9H), 0.91 (dd, J = 7.3, 3.0 Hz, 6H), 0.89 (s, 9H), 0.89–0.83 (m, 2H), 0.18 (s, 3H), 0.15 (s, 3H), 0.13 (s, 3H), 0.12 (s, 3H), 0.05 (s, 9H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 201.9, 147.3, 134.0, 118.0, 112.7, 106.6, 88.1, 83.0, 82.0, 81.2, 77.0, 75.0, 67.3, 60.1, 49.2, 43.6, 33.6, 28.7, 26.7, 26.5, 26.1, 24.7, 21.8, 18.5, 18.1, 17.2, 17.2, 17.2, 17.0, 14.2, 13.4, 2.5, −1.9, −2.0, −3.9, −4.3. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_42_H_82_O_8_Si_4_Na, 849.4979; found, 849.4980. Specific rotation: [α]D^20^ −24.5 (c 0.50, CH_2_Cl_2_).
Compound 63
To a stirred solution of the ketone 4 (140 mg, 193 μmol, 1 equiv) in dry DCM (4 mL) was added trimethylsilyl cyanide (192 mg, 242 μL, 1.93 mmol, 10 equiv). The resulting clear solution was then chilled to −15 °C, before titanium tetrachloride (1 M in DCM, 580 μL, 580 μmol, 3 equiv) was added dropwise. The now yellow reaction mixture was stirred for 1 h at −15 °C until TLC had indicated complete conversion. Then, sat. aqueous NaHCO_3_ solution was added to quench the reaction and the aqueous phase was extracted twice with DCM. The combined organic layers were dried over Na_2_SO_4_, filtered, and concentrated. The residue was purified via flash column chromatography (toluene/ethyl acetate, 150:1) to give 93 mg (64%) of the cyanohydrin 63 as colorless oil. ^1^H NMR (600 MHz, CDCl_3_): δ 5.90 (ddt, J = 17.4, 10.3, 7.3 Hz, 1H), 5.41 (s, 1H), 5.27 (s, 1H), 5.11–5.03 (m, 2H), 4.83–4.75 (m, 2H), 4.24 (dd, J = 10.3, 2.2 Hz, 1H), 4.07 (dd, J = 2.8, 1.0 Hz, 1H), 3.39 (s, 1H), 3.03 (dd, J = 10.7, 2.8 Hz, 1H), 2.74 (dd, J = 15.6, 1.1 Hz, 1H), 2.38–2.27 (m, 3H), 1.91–1.84 (m, 1H), 1.70 (ddd, J = 13.1, 10.8, 2.2 Hz, 1H), 1.41 (s, 3H), 1.39 (s, 3H), 1.31 (s, 3H), 1.11 (s, 3H), 1.03 (dd, J = 7.2, 1.0 Hz, 6H), 0.95 (dd, J = 7.2, 4.0 Hz, 6H), 0.93 (s, 9H), 0.90–0.82 (m, 2H), 0.86 (s, 9H), 0.23 (s, 3H), 0.15 (s, 3H), 0.15 (s, 3H), 0.11 (s, 3H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 146.3, 134.1, 120.8, 118.0, 114.2, 106.7, 82.9, 81.9, 81.4, 76.9, 73.7, 73.0, 66.8, 58.7, 55.1, 43.8, 33.1, 28.9, 27.0, 26.3, 25.9, 24.5, 21.9, 18.3, 18.0, 17.1, 16.9, 16.9, 16.8, 13.6, 13.1, −2.1, −2.4, −3.9, −4.5. HRMS (ESI) m/z: [M + H]^+^ calcd for C_39_H_74_NO_7_Si_3_, 752.4768; found, 752.4767. Specific rotation: [α]D^20^ +23.0 (c 0.50, CH_2_Cl_2_).
Compound 64
To a stirred solution of the starting material 63 (93 mg, 124 μmol, 1 equiv) in dry toluene (2 mL) was added DIBAL-H (1 M in hexane, 620 μL, 620 μmol, 5 equiv) at −80 °C. The resulting clear mixture was then stirred at this temperature for 2 h. At this point, TLC had indicated full conversion. The reaction was quenched by the addition of water (2 M in THF) and subsequently with sat. aqueous NH_4_Cl solution, before it was allowed to reach room temperature. It was then vigorously stirred for 1 h at room temperature, before the aqueous phase was extracted twice with toluene. The combined organic layers were filtered over a short plug of silica and concentrated subsequently. (If the filtration was omitted, partial destruction of the aldehyde was observed on TLC). The residue was purified via column chromatography (petroleum ether/ethyl acetate, 30:1) to afford 82 mg (88%) of the aldehyde 64 as colorless oil. ^1^H NMR (600 MHz, CDCl_3_): δ 9.30 (s, 1H), 5.88 (ddt, J = 17.4, 10.2, 7.3 Hz, 1H), 5.25 (s, 1H), 5.10–5.02 (m, 3H), 4.79 (d, J = 10.7 Hz, 1H), 4.50–4.45 (m, 1H), 4.17 (dd, J = 9.7, 2.4 Hz, 1H), 4.13 (d, J = 2.7 Hz, 1H), 3.66 (s, 1H), 3.15 (dd, J = 10.8, 2.8 Hz, 1H), 2.37–2.27 (m, 2H), 2.24 (dd, J = 15.1, 0.8 Hz, 1H), 1.98 (d, J = 15.2 Hz, 1H), 1.63–1.50 (m, 2H), 1.43 (s, 3H), 1.42 (s, 3H), 1.31 (s, 3H), 1.10 (s, 3H), 1.03 (dd, J = 7.3, 3.0 Hz, 6H), 0.96 (s, 9H), 0.95–0.89 (m, 7H), 0.89–0.82 (m, 1H), 0.88 (s, 9H), 0.23 (s, 3H), 0.15 (s, 3H), 0.12 (s, 3H), 0.10 (s, 3H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 202.9, 147.7, 134.0, 118.1, 113.4, 106.7, 85.3, 83.9, 82.4, 81.4, 77.2, 73.8, 66.8, 55.4, 54.9, 43.8, 33.4, 28.9, 26.9, 26.3, 26.1, 24.9, 21.9, 18.4, 18.1, 17.1, 16.9, 16.9, 16.8, 13.6, 13.1, −2.0, −2.1, −3.8, −4.3. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_39_H_74_O_8_Si_3_Na, 777.4583; found, 777.4580. Specific rotation: [α]D^20^ +33.6 (c 0.50, CH_2_Cl_2_).
Compound 66
The aldehyde 64 (55 mg, 72.8 μmol, 1 equiv) was dissolved in ether (1.5 mL). Then, water (500 μL) was added, followed by tetrabutylammonium iodide (13 mg, 36.4 μmol, 0.5 equiv) and cis-crotyltrifluoroborate 65 (24 mg, 146 μmol, 2 equiv). The resulting biphasic mixture was stirred vigorously for 12 h. As TLC indicated incomplete conversion, further cis-crotyltrifluoroborate 65 (24 mg, 146 μmol, 2 equiv) was added, which pushed the reaction to full completion after 6 h. Subsequently, the aqueous phase was extracted twice with ether, the combined organic layers were dried over Na_2_SO_4_, filtered, and concentrated. The obtained residue was purified via column chromatography (petroleum ether/ether, 25:1) to yield 54 mg (91%) of the 1,2-diol 66 as colorless oil. ^1^H NMR (600 MHz, CDCl_3_): δ 5.87 (ddt, J = 17.6, 10.4, 7.4 Hz, 1H), 5.81 (ddd, J = 17.4, 10.4, 7.2 Hz, 1H), 5.22 (s, 1H), 5.16–5.06 (m, 3H), 4.98–4.88 (m, 2H), 4.74 (dd, J = 8.8, 2.3 Hz, 1H), 4.71 (d, J = 10.3 Hz, 1H), 4.21–4.16 (m, 1H), 4.06 (dd, J = 3.0, 0.9 Hz, 1H), 3.58 (dd, J = 5.8, 2.9 Hz, 1H), 3.38 (s, 1H), 2.96 (dd, J = 10.3, 3.0 Hz, 1H), 2.63 (dd, J = 15.0, 1.1 Hz, 1H), 2.51 (d, J = 5.8 Hz, 1H), 2.49–2.43 (m, 1H), 2.39–2.27 (m, 2H), 1.76–1.65 (m, 3H), 1.40 (s, 3H), 1.40 (s, 3H), 1.31 (s, 3H), 1.09 (s, 3H), 1.02 (dd, J = 7.2, 3.9 Hz, 6H), 0.95 (s, 9H), 0.94–0.91 (m, 9H), 0.85 (s, 9H), 0.85–0.79 (m, 2H), 0.22 (s, 3H), 0.14 (s, 3H), 0.12 (s, 3H), 0.10 (s, 3H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 149.4, 144.6, 133.5, 118.5, 112.8, 112.8, 106.9, 84.4, 84.3, 81.5, 81.3, 78.6, 76.1, 72.2, 67.0, 52.4, 51.3, 43.7, 38.0, 33.9, 28.6, 26.8, 26.4, 26.4, 26.2, 21.6, 18.4, 18.2, 17.2, 17.1, 17.1, 17.0, 14.0, 13.6, 13.2, −2.0, −2.4, −3.7, −4.6. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_43_H_82_O_8_Si_3_Na, 833.5209; found, 833.5209. Specific rotation: [α]D^20^ +107.3 (c 0.50, CH_2_Cl_2_).
Compound 68
To a stirred solution of the 1,2-diol 66 (8 mg, 9.9 μmol, 1 equiv) in dry DCM (500 μL) was added trichloroacetyl isocyanate 67 (0.5 M in DCM, 30 μL, 14.8 μmol, 1.5 equiv) at room temperature. After 5 min of stirring, TLC had indicated complete conversion. The reaction was then quenched by the addition of water; the resulting mixture was extracted with DCM twice. The combined organic layers were dried over Na_2_SO_4_ and concentrated. The obtained crude mixture was subjected to flash column chromatography (petroleum ether/ether, 25:1) to give 10 mg (quant.) of the carbamate 68 as a white solid. ^1^H NMR (600 MHz, CDCl_3_): δ 8.08 (s, 1H), 5.91 (ddt, J = 17.5, 10.3, 7.3 Hz, 1H), 5.82 (ddd, J = 17.1, 10.5, 6.6 Hz, 1H), 5.35 (s, 1H), 5.34 (d, J = 2.8 Hz, 1H), 5.30 (s, 1H), 5.11–5.01 (m, 2H), 4.98–4.92 (m, 3H), 4.58 (d, J = 10.8 Hz, 1H), 4.22 (m, 2H), 3.95 (s, 1H), 2.75 (dddd, J = 10.1, 8.7, 4.4, 2.9 Hz, 1H), 2.64 (dd, J = 10.6, 2.9 Hz, 1H), 2.61 (dd, J = 15.1, 1.1 Hz, 1H), 2.39 (ddt, J = 14.0, 7.2, 1.3 Hz, 1H), 2.30 (ddt, J = 14.0, 7.5, 1.2 Hz, 1H), 2.01 (ddd, J = 13.3, 9.1, 1.9 Hz, 1H), 1.91 (d, J = 15.2 Hz, 1H), 1.70 (ddd, J = 13.6, 10.9, 3.3 Hz, 1H), 1.44 (s, 3H), 1.39 (s, 3H), 1.29 (s, 3H), 1.18 (s, 3H), 1.06–0.99 (m, 9H), 0.96 (s, 9H), 0.95 (dd, J = 7.3, 2.4 Hz, 6H), 0.90–0.85 (m, 2H), 0.84 (s, 9H), 0.23 (s, 3H), 0.17 (s, 3H), 0.12 (s, 3H), 0.10 (s, 3H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 156.9, 149.2, 149.1, 142.3, 134.3, 117.8, 114.2, 114.2, 106.3, 92.1, 84.5, 82.3, 81.6, 80.8, 79.0, 77.8, 75.1, 67.0, 54.8, 51.2, 43.5, 38.2, 33.0, 28.8, 26.8, 26.6, 26.3, 26.2, 22.1, 18.6, 18.1, 17.2, 17.0, 17.0, 17.0, 14.2, 13.7, 13.2, −1.6, −1.9, −3.7, −3.9. HRMS (ESI) m/z: [M
- Na]^+^ calcd for C_46_H_82_Cl_3_NO_10_Si_3_Na, 1020.4204; found, 1020.4206. Specific rotation: [α]D^20^ −3.7 (c 0.25, CH_2_Cl_2_). Melting point: mp 66.9–69.2 °C.
Compound 70
The 1,2-diol 66 (53 mg, 65.3 μmol, 1 equiv) was dissolved in DCE/DMSO (750 μL each). Then, IBX (91 mg, 327 μmol, 5 equiv) was added in one portion. The obtained clear solution was heated to 55 °C (oil bath) and stirred for 12 h, until TLC had indicated complete conversion. The suspension was then directly filtered over silica (5 g) and eluted with DCM. The product containing fractions were combined, and the solvents were distilled off. The residue was purified via column chromatography (petroleum ether/ether, 25:1) to give 47 mg (89%) of the ketone 70 as colorless oil. ^1^H NMR (600 MHz, CDCl_3_): δ 5.97 (ddd, J = 17.4, 10.4, 8.2 Hz, 1H), 5.89 (ddt, J = 17.5, 10.2, 7.3 Hz, 1H), 5.16 (s, 1H), 5.11–5.01 (m, 2H), 5.03–4.90 (m, 2H), 4.89 (s, 1H), 4.71 (d, J = 10.7 Hz, 1H), 4.49 (dd, J = 9.9, 3.3 Hz, 1H), 4.17–4.14 (m, 1H), 4.13 (d, J = 2.8 Hz, 1H), 3.89 (s, 1H), 3.69 (dtd, J = 8.2, 7.2, 6.2 Hz, 1H), 3.39 (dd, J = 10.8, 2.8 Hz, 1H), 2.31 (qdt, J = 13.9, 7.4, 1.2 Hz, 2H), 2.22–2.15 (m, 1H), 1.99 (d, J = 14.9 Hz, 1H), 1.50 (ddd, J = 10.3, 6.1, 3.2 Hz, 2H), 1.46 (s, 3H), 1.40 (s, 3H), 1.32 (s, 3H), 1.11 (d, J = 7.2 Hz, 3H), 1.09 (s, 3H), 1.03 (d, J = 7.2 Hz, 6H), 0.97 (s, 9H), 0.92 (dd, J = 7.2, 6.3 Hz, 6H), 0.89 (s, 9H), 0.86–0.78 (m, 2H), 0.23 (s, 3H), 0.16 (s, 3H), 0.09 (s, 3H), 0.08 (s, 3H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 213.8, 147.9, 138.9, 134.2, 117.9, 114.6, 112.0, 106.7, 87.0, 84.0, 82.7, 81.5, 74.4, 77.1, 66.5, 59.7, 55.4, 45.1, 43.8, 32.8, 29.0, 27.0, 26.4, 26.0, 25.8, 21.8, 18.4, 18.1, 17.6, 17.1, 16.9, 16.9, 16.9, 13.6, 13.1, −2.0, −2.2, −3.8, −4.4. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_43_H_80_O_8_Si_3_Na, 831.5053; found, 831.5051. Specific rotation: [α]D^20^ +38.9 (c 0.50, CH_2_Cl_2_).
Compound 3
To a stirred solution of the ketone 70 (30 mg, 37.1 μmol, 1 equiv) were added acetic anhydride (0.5 M in toluene, 150 μL, 75 μmol, 2 equiv) and trimethylsilyl trifluoromethanesulfonate (0.1 M in toluene, 75 μL, 7.5 μmol, 0.2 equiv) sequentially at room temperature. As soon as TLC had indicated complete conversion (10 min), the reaction was quenched with sat. aqueous NaHCO_3_ solution. The aqueous phase was extracted twice with toluene, and the combined organic phases were dried over Na_2_SO_4_, filtered, and concentrated. The obtained residue was purified via flash column chromatography (petroleum ether/ether, 15:1) to afford 25 mg (79%) of the acetylated product 3 as colorless oil. ^1^H NMR (600 MHz, CDCl_3_): δ 5.86 (ddt, J = 17.4, 10.2, 7.3 Hz, 1H), 5.74 (ddd, J = 17.6, 10.3, 7.7 Hz, 1H), 5.23 (s, 1H), 5.19–5.02 (m, 5H), 4.99 (d, J = 10.2 Hz, 1H), 4.44–4.40 (m, 1H), 4.24–4.15 (m, 3H), 3.02 (dd, J = 16.1, 1.3 Hz, 1H), 2.84 (dd, J = 10.2, 3.1 Hz, 1H), 2.35–2.26 (m, 2H), 2.24 (d, J = 16.1 Hz, 1H), 2.03 (s, 3H), 1.63–1.53 (m, 2H), 1.44 (s, 3H), 1.32 (s, 3H), 1.26 (s, 3H), 1.14 (d, J = 6.7 Hz, 3H), 1.06–1.02 (m, 9H), 0.97–0.92 (m, 6H), 0.94 (s, 9H), 0.92 (s, 9H), 0.90–0.80 (m, 2H), 0.19 (s, 3H), 0.19 (s, 3H), 0.18 (s, 3H), 0.12 (s, 3H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 205.4, 169.3, 145.1, 139.6, 133.9, 118.1, 116.1, 114.5, 106.5, 91.5, 83.2, 81.3, 81.2, 77.1, 74.5, 66.4, 52.5, 51.5, 43.7, 42.6, 32.6, 28.5, 26.7, 26.2, 26.2, 24.5, 21.9, 21.3, 19.2, 18.5, 18.4, 17.2, 17.0, 16.9, 16.9, 13.7, 13.2, −1.5, −1.9, −3.1, −4.0. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_45_H_82_O_9_Si_3_Na, 873.5159; found, 873.5161. Specific rotation: [α]D^20^ +27.0 (c 0.50, CH_2_Cl_2_).
Compound 75
To a flask containing the freshly prepared trifluoroborate 74 (21 mg, 90.0 μmol, 4 equiv), the aldehyde 64 (17 mg, 22.5 μmol, 1 equiv) dissolved in ether (1 mL) was added. Then, water (500 μL) was added, followed by tetrabutylammonium iodide (4 mg, 11.0 μmol, 0.5 equiv). The resulting biphasic mixture was stirred vigorously for 30 min, until TLC had indicated complete conversion. Subsequently, the aqueous phase was extracted twice with ether, the combined organic layers were dried over Na_2_SO_4_, filtered, and concentrated. The obtained residue was purified via column chromatography (petroleum ether/ether, 25:1) to yield 11 mg (56%) of the relay precursor 75 as colorless oil. ^1^H NMR (600 MHz, CDCl_3_): δ 5.88 (ddt, J = 17.5, 10.3, 7.4 Hz, 1H), 5.81 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 5.46 (ddt, J = 15.4, 8.8, 1.3 Hz, 1H), 5.35 (dt, J = 15.4, 6.6 Hz, 1H), 5.22 (s, 1H), 5.15–5.04 (m, 3H), 5.01–4.90 (m, 2H), 4.80 (d, J = 9.6 Hz, 1H), 4.71 (d, J = 10.3 Hz, 1H), 4.20 (dd, J = 10.2, 1.9 Hz, 1H), 4.07–4.03 (m, 1H), 3.42 (s, 1H), 3.38 (dd, J = 5.9, 2.7 Hz, 1H), 2.96 (dd, J = 10.3, 3.0 Hz, 1H), 2.65 (d, J = 5.9 Hz, 1H), 2.49 (dd, J = 15.4, 1.0 Hz, 1H), 2.44 (ddd, J = 9.2, 7.0, 2.6 Hz, 1H), 2.39–2.28 (m, 2H), 2.07–2.02 (m, 2H), 1.98 (tdd, J = 7.3, 5.1, 3.7 Hz, 2H), 1.79–1.67 (m, 2H), 1.59 (d, J = 15.5 Hz, 1H), 1.47–1.43 (m, 2H), 1.43 (s, 3H), 1.36 (s, 3H), 1.33 (s, 3H), 1.10 (s, 3H), 1.02 (dd, J = 7.2, 3.8 Hz, 6H), 0.95 (s, 9H), 0.94–0.91 (m, 9H), 0.85 (s, 9H), 0.86–0.79 (m, 2H), 0.22 (s, 3H), 0.13 (s, 3H), 0.10 (s, 3H), 0.08 (s, 3H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 149.6, 139.1, 133.5, 133.4, 129.7, 118.5, 114.4, 112.7, 106.9, 84.8, 84.7, 81.4, 81.3, 78.7, 76.1, 74.1, 67.0, 53.0, 51.0, 43.6, 37.9, 34.1, 33.6, 32.5, 28.8, 28.7, 26.8, 26.5, 26.5, 26.2, 21.6, 20.9, 18.4, 18.2, 17.2, 17.1, 17.1, 17.0, 13.9, 13.2, −2.0, −2.4, −3.7, −4.6. HRMS (ESI) m/z: [M + H]^+^ calcd for C_48_H_91_O_8_Si_3_, 879.6016; found, 879.6015. Specific rotation: [α]D^20^ +11.1 (c 0.25, CH_2_Cl_2_).
Compound 77
The aldehyde 64 (60 mg, 79.4 μmol, 1 equiv) was dissolved in ether (1.5 mL). Then, water (500 μL) was added, followed by tetrabutylammonium iodide (15 mg, 39.7 μmol, 0.5 equiv) and allyltrifluoroborate 76 (47 mg, 318 μmol, 4 equiv). The resulting biphasic mixture was stirred vigorously until TLC had indicated complete conversion after 2 h. Subsequently, the aqueous phase was extracted twice with ether, the combined organic layers were dried over Na_2_SO_4_, filtered, and concentrated. The obtained residue was purified via column chromatography (petroleum ether/ether, 25:1) to yield 57 mg (90%) of the 1,2-diol 77 as colorless oil. ^1^H NMR (600 MHz, CDCl_3_): δ 5.87 (ddt, J = 7.4, 10.2, 17.4 Hz, 1H), 5.78 (dddd, J = 6.3, 7.5, 10.2, 16.6 Hz, 1H), 5.23 (s, 1H), 5.12 (s, 1H), 5.11–4.97 (m, 4H), 4.87–4.82 (m, 1H), 4.71 (d, J = 10.3 Hz, 1H), 4.18 (dd, J = 2.3, 9.7 Hz, 1H), 4.13–4.10 (m, 1H), 3.54 (s, 1H), 3.49 (ddd, J = 1.9, 5.1, 10.3 Hz, 1H), 3.02 (dd, J = 3.0, 10.3 Hz, 1H), 2.72 (d, J = 5.1 Hz, 1H), 2.50–2.44 (m, 1H), 2.42–2.38 (m, 1H), 2.38–2.27 (m, 2H), 1.84–1.76 (m, 1H), 1.76–1.66 (m, 3H), 1.40 (s, 3H), 1.39 (s, 3H), 1.31 (s, 3H), 1.09 (s, 3H), 1.02 (dd, J = 3.2, 7.2 Hz, 6H), 0.95 (s, 9H), 0.93 (dd, J = 6.1, 7.3 Hz, 6H), 0.85 (s, 9H), 0.88–0.80 (m, 2H), 0.23 (s, 3H), 0.15 (s, 3H), 0.12 (s, 3H), 0.10 (s, 3H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 149.5, 137.1, 133.6, 118.5, 116.6, 113.0, 106.9, 85.4, 84.3, 81.4, 81.0, 78.6, 75.9, 70.3, 67.0, 51.6, 50.1, 43.6, 36.2, 34.1, 28.6, 26.8, 26.4, 26.4, 26.1, 21.6, 18.4, 18.2, 17.2, 17.1, 17.1, 17.0, 14.0, 13.2, −2.0, −2.3, −3.7, −4.6. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_42_H_80_O_8_Si_3_Na, 819.5053; found, 819.5057. Specific rotation: [α]D^20^ +93.4 (c 0.50, CH_2_Cl_2_).
Compound 78
The 1,2-diol 77 (30 mg, 37.6 μmol, 1 equiv) was dissolved in DCE/DMSO (750 μL each). Then, IBX (53 mg, 188 μmol, 5 equiv) was added in one portion. The obtained clear solution was heated to 55 °C (oil bath) and stirred for 12 h, until TLC had indicated complete conversion. The suspension was then directly filtered over silica (5 g) and eluted with DCM. The product containing fractions were combined, and the solvents were distilled off. The residue was purified via column chromatography (petroleum ether/ether, 25:1) to give 26 mg (85%) of the ketone 78 as colorless oil. ^1^H NMR (600 MHz, CDCl_3_): δ 5.89 (dddt, J = 3.3, 6.8, 10.8, 17.3 Hz, 2H), 5.17 (s, 1H), 5.12–5.01 (m, 4H), 4.92 (s, 1H), 4.76 (d, J = 10.7 Hz, 1H), 4.59–4.54 (m, 1H), 4.18 (dd, J = 2.9, 9.6 Hz, 1H), 4.16 (d, J = 2.8 Hz, 1H), 3.89 (s, 1H), 3.39 (ddt, J = 1.4, 7.1, 18.1 Hz, 1H), 3.33 (dd, J = 2.9, 10.7 Hz, 1H), 3.11 (ddt, J = 1.5, 6.6, 18.1 Hz, 1H), 2.38–2.27 (m, 2H), 2.16 (d, J = 15.0 Hz, 1H), 1.96 (d, J = 15.0 Hz, 1H), 1.61–1.48 (m, 2H), 1.44 (s, 3H), 1.41 (s, 3H), 1.32 (s, 3H), 1.10 (s, 3H), 1.04 (d, J = 7.3 Hz, 6H), 0.97 (s, 9H), 0.92 (dd, J = 5.9, 7.3 Hz, 6H), 0.89 (s, 9H), 0.88–0.81 (m, 2H), 0.23 (s, 3H), 0.16 (s, 3H), 0.09 (s, 6H). ^13^C{^1^H} NMR (151 MHz, CDCl_3_): δ 210.2, 148.3, 134.1, 131.4, 118.0, 117.9, 111.9, 106.7, 87.0, 83.8, 82.6, 81.5, 77.5, 74.3, 66.6, 58.2, 54.5, 43.8, 42.5, 33.0, 28.9, 26.9, 26.4, 26.1, 25.6, 21.8, 18.4, 18.1, 17.1, 16.9, 16.9, 16.9, 13.6, 13.1, −2.0, −2.1, −3.8, −4.4. HRMS (ESI) m/z: [M + H]^+^ calcd for C_42_H_79_O_8_Si_3_, 795.5077; found, 795.5077. Specific rotation: [α]D^20^ +45.6 (c 0.50, CH_2_Cl_2_).
Compound 79
The starting material 78 (17 mg, 21.0 μmol, 1 equiv) was dissolved in dry DCE (2.5 mL) before the reaction mixture was degassed via freeze–pump–thaw cycles (3×). Then, second-generation Grubbs–Hoveyda catalyst [301224-40-8] (0.1 M in degassed DCE, 40 μL, 4 μmol, 0.2 equiv) was added dropwise. The resulting green solution was heated to 65 °C (oil bath) and stirred for 3 h at the respective temperature. As the subsequent TLC indicated incomplete conversion, more catalyst (0.1 M in degassed DCE, 40 μL, 4 μmol, 0.2 equiv) was added, which pushed the reaction to full completion after another 2 h. Then, the reaction mixture was exposed to air, to oxidize the remaining catalyst, before the solvent was distilled off. Crude brown oil was obtained, which was purified via column chromatography (petroleum ether/ether, 20:1) to yield 10 mg (61%) of the macrocycle 79. ^1^H NMR ((Z)-isomer, 600 MHz, CDCl_3_): δ 5.82–5.71 (m, 2H), 5.34 (s, 1H), 5.15 (s, 1H), 4.95 (d, J = 10.7 Hz, 1H), 4.39 (d, J = 10.6 Hz, 1H), 4.24 (d, J = 2.9 Hz, 1H), 3.98 (s, 1H), 3.54 (dd, J = 1.8, 10.7 Hz, 1H), 3.40 (dd, J = 10.3, 19.8 Hz, 1H), 3.33–3.26 (m, 1H), 3.14 (dd, J = 3.0, 10.8 Hz, 1H), 2.36–2.23 (m, 3H), 2.17–2.04 (m, 3H), 1.47 (s, 3H), 1.41 (s, 3H), 1.31 (s, 3H), 1.27 (s, 3H), 1.07 (dd, J = 5.2, 7.3 Hz, 6H), 0.96 (s, 9H), 0.94–0.91 (m, 6H), 0.90 (s, 9H), 0.89–0.82 (m, 2H), 0.19 (s, 3H), 0.18 (s, 3H), 0.15 (s, 3H), 0.14 (s, 3H). ^13^C{^1^H} NMR ((Z)-isomer, 151 MHz, CDCl_3_): δ 209.5, 144.5, 128.2, 123.1, 116.4, 106.3, 85.0, 83.3, 82.6, 82.0, 74.5, 73.9, 67.3, 57.9, 54.5, 35.8, 35.5, 33.8, 28.9, 26.7, 26.3, 26.2, 24.9, 24.7, 18.5, 18.3, 17.1, 17.0, 17.0, 16.9, 13.4, 13.3, −1.7, −1.7, −3.5, −4.0. HRMS (ESI) m/z: [M + Na]^+^ calcd for C_40_H_74_O_8_Si_3_Na, 789.4583; found, 789.4587. Specific rotation: [α]D^20^ +8.3 (c 0.25, CH_2_Cl_2_).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hohmann J.; Evanics F.; Dombi G.; Molnár J.; SzabóP. Euphosalicin, a new diterpene polyester with multidrug resistance reversing activity from Euphorbia salicifolia. Tetrahedron 2001, 57, 211–215. 10.1016/S 0040-4020(00)00982-0. · doi ↗
- 2Kupchan S. M.; Sigel C. W.; Matz M. J.; Saenz Renauld J. A.; Haltiwanger R. C.; Bryan R. F. Jatrophone, a novel macrocyclic diterpenoid tumor inhibitor from Jatropha gossypiifolia. J. Am. Chem. Soc. 1970, 92 (14), 4476–4477. 10.1021/ja 00717 a 066. · doi ↗
- 3Duarte N.; Lage H.; Ferreira M. J. Three New Jatrophane Polyesters and Antiproliferative Constituents from Euphorbia tuckeyana. Planta Med. 2008, 74, 61–68. 10.1055/s-2007-993765.18176909 · doi ↗ · pubmed ↗
- 4Yang H.; Mamatjan A.; Tang D.; Aisa H. A. Jatrophane diterpenoids as multidrug resistance modulators from Euphorbia sororia. Bioorg. Chem. 2021, 112, 10498910.1016/j.bioorg.2021.104989.34022709 · doi ↗ · pubmed ↗
- 5a Smith A. B.; Guaciaro M. A.; Schow S. R.; Wovkulich P. M.; Toder B. H.; Hall T. W. A Strategy for the Total Synthesis of Jatrophone: Synthesis of Normethyljatrophone. J. Am. Chem. Soc. 1981, 103 (1), 219–222. 10.1021/ja 00391 a 054. · doi ↗
- 6a Matsuura T.; Nishiyama S.; Yamamura S. Efficient Synthesis of the Optically Active Cyclopentane Derivative, A Versatile Intermediate Toward the Euphorbia Diterpenes. Chem. Lett. 1993, 22, 1503–1504. 10.1246/cl.1993.1503. · doi ↗
- 7Rinner U.; Lentsch C.; Fürst R. Enyne Metathesis Approach towards the Cyclopentane Motif of Jatrophane Diterpenes. Synlett 2013, 24 (20), 2665–2670. 10.1055/s-0033-1339923. · doi ↗
- 8Miller J. A.; Negishi E. Zirconium-catalyzed allylalumination and benzylalumination of alkynes. Tetrahedron Lett. 1984, 25, 5863–5866. 10.1016/S 0040-4039(01)81705-6. · doi ↗