Protocol to identify defined reprogramming factor expression using a factor-indexing single-nuclei multiome sequencing approach
Liangru Fei, Kaiyang Zhang, Sampsa Hautaniemi, Biswajyoti Sahu

TL;DR
This paper introduces a protocol using a new sequencing method to track specific gene activity during cell reprogramming.
Contribution
A novel factor-indexing single-nuclei multiome sequencing approach for tracking reprogramming factors.
Findings
Barcoding transcription factors allows tracking of their expression in reprogramming cells.
Human fibroblasts can be reprogrammed into pancreatic ductal-like cells using defined factors.
FI-snMultiome-seq enables analysis of heterogeneous reprogramming cell populations.
Abstract
Ectopic expression of lineage-specific transcription factors (TFs) of another cell type can induce cell fate reprogramming. However, the heterogeneity of reprogramming cells has been a challenge for data interpretation and model evaluation. Here, we present a protocol to characterize cells expressing defined factors during direct cell reprogramming using a factor-indexing approach based on single-nuclei multiome sequencing (FI-snMultiome-seq). We describe the steps for barcoding TFs, converting human fibroblasts to pancreatic ductal-like cells using defined TFs, and preparing library for FI-snMultiome-seq analysis. For complete details on the use and execution of this protocol, please refer to Fei et al.1 •Generating barcoded Gateway destination vectors by PCR•Barcoding individual transcription factor constructs by Gateway cloning•Direct reprogramming of pancreatic ductal-like cells…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
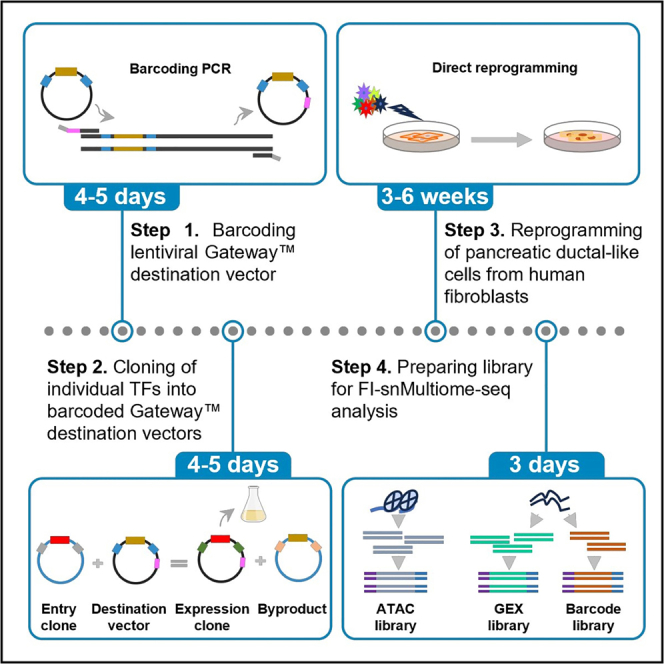 Figure 1
Figure 1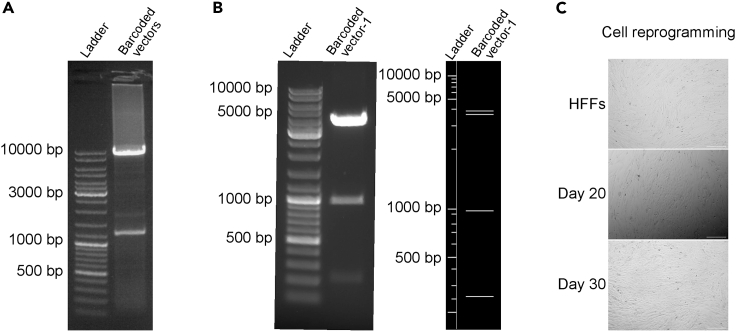 Figure 2
Figure 2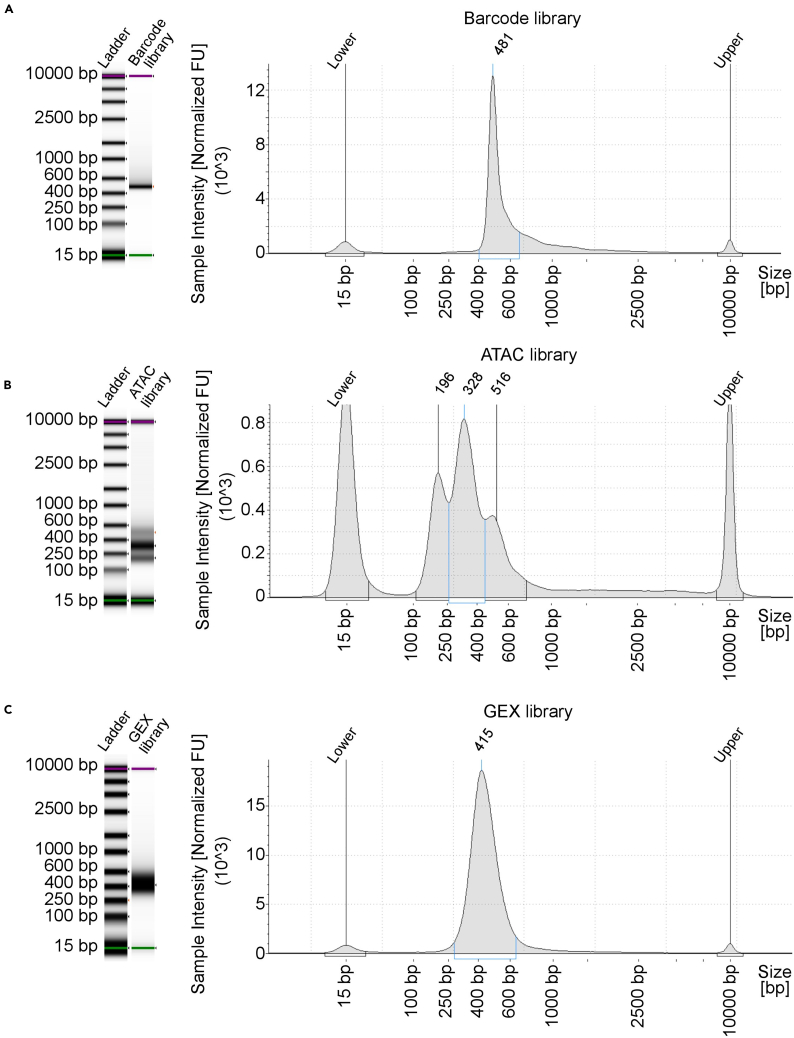 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSingle-cell and spatial transcriptomics · Advanced biosensing and bioanalysis techniques · Renal and related cancers
Before you begin
This protocol below describes the steps to generate barcoded TF constructs for inducing pancreatic ductal-like cells from human fibroblasts and for further use with FI-snMultiome-seq analysis. Using this protocol, we identified the cells carrying different combinations of reprogramming TFs from a six-TF (6F) pool and compared the enrichment of ductal cell signatures in each clone. We demonstrated that all six TFs are necessary and sufficient for efficient conversion of pancreatic ductal-like cells from human fibroblasts.1 Gene expression and chromatin profiling of the cells using FI-snMultiome-seq assay provided a high-resolution single-nucleus analysis of TF-mediated reprogramming during transdifferentiation.
The protocol is versatile and can be adapted to other direct reprogramming models and experimental settings that need to characterize defined cells expressing the gene(s) of interest from exogeneous vectors in a heterogeneous cell population.
Before starting, you need to identify candidate TFs required for your reprogramming model or the gene(s) of interest for your experimental setting, and clone the full-length open reading frames (ORF) of all individual TFs/genes into Gateway donor vectors. Also, verify to have all the reagents and equivalent equipment described in the protocol.
Institutional permissions
Experiments on live vertebrates or higher invertebrates must be performed in accordance with national guidelines and regulations. Experiments involving lentivirus must be conducted under Biosafety Level 2 (BSL2) as per institutional guidelines. We remind readers to obtain all the necessary permissions from the relevant institutions before starting the experiment.
Identify candidate TFs for your reprogramming model
Timing: variable
- 1.Select candidate TFs based on their reported role in developmental biology and/or use computational framework, such as Mogrify (https://mogrify.net/index),2 to predict candidate TFs required for transcriptomic switches from your source cell type to any target cell type.
Note: Computational frameworks based on gene expression data and regulatory network information can usually predict the TFs required for cell fate conversion for broad tissue types instead of specific cell types.2^,^3 You may combine the literature-curated TFs and computational framework-predicted TFs as initial pool to start with.
Clone TF ORFs into Gateway donor vectors to generate entry clones
Timing: 1–2 weeks
- 2.Obtain the full-length ORFs of all individual TFs on Gateway donor vectors from publicly available resources such as Addgene or get the attB flanked TF ORFs from any commercial source and clone into Gateway donor vector (e.g., pDNOR221) by BP Reaction following manufacturer’s instructions.
Key resources table
REAGENT or RESOURCESOURCEIDENTIFIERBacterial and virus strainsOne Shot Stbl3 chemically competent E. coliThermo Fisher ScientificCat# C737303One Shot ccdB Survival 2 T1R competent cellsThermo Fisher ScientificCat# A10460pLenti6/V5-Barcode-FOXA2Fei et al.1N/ApLenti6/V5-Barcode-HNF1BFei et al.1N/ApLenti6/V5-Barcode-HNF6Fei et al.1N/ApLenti6/V5-Barcode-PDX1Fei et al.1N/ApLenti6/V5-Barcode-SOX9Fei et al.1N/ApLenti6/V5-Barcode-SOX17Fei et al.1N/AChemicals, peptides, and recombinant proteinsAscorbic AcidSigma-AldrichCat# A1300000Retinoic AcidSigma-AldrichCat# R2625PD0325901Sigma-AldrichCat# PZ0162Poly-L-lysineSigma-AldrichCat# P4707PolybreneSigma-AldrichCat# S2667CHIR99021StemMACSCat# 130-106-539LDN193189StemMACSCat# 130-103-925Activin APeproTechCat# AF-120-14EFGF7PeproTechCat# AF-100-19Complete human epithelial cell mediumCell BiologicsCat# PB-H-6621SPRIselect beadsBeckman CoulterCat# B23318Electrophoresis gels, peqGOLD, universal agaroseVWRCat# 732-2789PBuffer EBQIAGENCat# 19086Lenti-X concentratorTakaraCat# 631232MatrigelCorningCat# 356230Q5 high-fidelity DNA polymeraseNew England BiolabsCat# M0491KAPA HiFi HotStart ReadyMixKapa BiosystemsCat# KK2602dNTP mixThermo Fisher ScientificCat# R0192SYBR Green I nucleic acid gel stainThermo Fisher ScientificCat# S7563Nuclease-free waterThermo Fisher ScientificCat# AM9937FastDigest NheIThermo Fisher ScientificCat# FD0974FastDigest DpnlThermo Fisher ScientificCat# FD1703FastDigest XhoIThermo Fisher ScientificCat# FD0694FastDigest AflIIThermo Fisher ScientificCat# FD0834T4 DNA ligaseThermo Fisher ScientificCat# EL0011Gateway LR Clonase II enzyme mixThermo Fisher ScientificCat# 11791020Lipofectamine 2000 transfection reagentThermo Fisher ScientificCat# 11668019Geneticin selective antibiotic (G418 sulfate)Thermo Fisher ScientificCat# 10131027GlutaMAX supplementThermo Fisher ScientificCat# 35050038B27 supplementThermo Fisher ScientificCat# 17504044Sodium pyruvateThermo Fisher ScientificCat# 11360070L-glutamineThermo Fisher ScientificCat# 25030024MEM non-essential amino acids (MEM NEAAs)Thermo Fisher ScientificCat# 11140035Penicillin-streptomycin (Pen-Strep)Thermo Fisher ScientificCat# 151401222-mercaptoethanolThermo Fisher ScientificCat# 21985023HEPESThermo Fisher ScientificCat# 15630080Insulin-transferrin-selenium-ethanolamine (ITS -X)Thermo Fisher ScientificCat# 51500056KnockOut serum replacementThermo Fisher ScientificCat# 10828010Fetal bovine serum (FBS)Thermo Fisher ScientificCat# 10270106StemPro Accutase cell dissociation reagentThermo Fisher ScientificCat# A1110501Opti-MEM I reduced serum medium (Opti-MEM)Thermo Fisher ScientificCat# 51985034Advanced DMEM/F-12Thermo Fisher ScientificCat# 12634010DMEM, high glucose, no glutamineThermo Fisher ScientificCat# 11960085DPBS, no calcium, no magnesiumThermo Fisher ScientificCat# 14190250Critical commercial assaysChromium next GEM single cell multiome ATAC + gene expression reagent bundle10× GenomicsCat# PN-1000285Dual index kit TT set A10× GenomicsCat# PN-100021NucleoSpin gel and PCR clean-up, mini kit for gel extraction and PCR clean-upMACHEREY-NAGELCat# 740609NucleoSpin plasmid, mini kit for plasmid DNAMACHEREY-NAGELCat# 740588NucleoBond Xtra midi EF, midi kit for endotoxin-free plasmid DNAMACHEREY-NAGELCat# 740420KAPA library quantification kitKapa BiosystemsCat# KK4854Agilent high sensitivity D5000 ScreenTapeAgilentCat# 5067-5592Agilent high sensitivity D5000 reagentsAgilentCat# 5067-5593Deposited dataRaw dataFei et al.1GEO: [GSE216859](GSE216859)ENCODE blacklisted genomic regions for hg38ENCODEENCFF356LFXHuman reference genome NCBI build 38, GRCh38Genome Reference Consortiumhttps://www.ncbi.nlm.nih.gov/assembly/GCF_000001405Experimental models: Cell linesHuman foreskin fibroblasts (HFF)ATCCCat# CRL-2429293FT cell lineThermo Fisher ScientificCat# R70007OligonucleotidesBarcode_Temp_Nhel_Forward primer: CATGCTAGCNNNNNNNNNNNNNNNNNNNNCGAGCTCGGTACCTTTAAGACCFei et al.1N/ABarcode_Temp_Nhel_Reverse primer: CATGCTAGCTTGTGCTTAGCCCTCCCACACFei et al.1N/ASseq_ORF sequencing primer: CCAGTGTGGTGGAATTCTGCAFei et al.1N/ASseq_Barcode sequencing primer: GCTGCAATAAACAAGTTCCTCTCACFei et al.1N/ATruseq_Read2_Vec_Amp_Forward primer: GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTAGGGCTAAGCACAAGCTAG∗CFei et al.1N/ATruseq_Read1_Amp_Reverse primer: ACACTCTTTCCCTACACGACGCTCTTCCGATC∗TFei et al.1N/ARecombinant DNApsPAX2AddgeneRRID: Addgene_12260pMD2.GAddgeneRRID: Addgene_12259pDONR221 vectorThermo Fisher ScientificCat# 12536017pLenti6/V5-DESTThermo Fisher ScientificCat# V49610Software and algorithmsSegIORoberts et al.4https://github.com/google/seqioBiopythonCock et al.5https://biopython.org/Seurat (v4.1.1)Hao et al.6https://satijalab.org/seurat/Signac (v1.7.0)Stuart et al.7https://stuartlab.org/signac/MACS2 (v2.2.7.1)Zhang et al.8http://github.com/taoliu/MACS/chromVAR (v1.18.0)Schep et al.9https://stuartlab.org/signac/reference/runchromvarScType (v1.0)Ianevski et al.10https://github.com/IanevskiAleksandr/sc-typeOtherMillipore Steriflip vacuum tube top filterSigmaCat# SE1M003M0010× magnetic separator10× GenomicsCat# 120250Agilent TapeStation 4150 systemAgilentCat# G2992AALightCycler 480RocheCat# 05015278001C1000 Touch thermal cyclerBio-RadCat# 1851197ThermoMixerEppendorfCat# 2231001005
Materials and equipment
Complete medium for 293FT cellsReagentFinal concentrationAmountMEM NEAAs (100×)1×5 mLL-Glutamine (200 mM)6 mM15 mLSodium Pyruvate (100 mM)1 mM5 mLFBS10%50 mLDMEMN/A425 mLTotalN/A500 mLStore at 4°C for up to 1 month. Serum reduced medium for 293FT cellsReagentFinal concentrationAmountFBS5%12.5 mLOpti-MEMN/A237.5 mLTotalN/A250 mLStore at 4°C for up to 1 month. Complete medium for fibroblastsReagentFinal concentrationAmountPen-Strep (10,000 U/mL)100 U/mL5 mLL-Glutamine (200 mM)2 mM5 mLSodium Pyruvate (100 mM)1 mM5 mLFBS10%50 mLDMEMN/A435 mLTotalN/A500 mLStore at 4°C for up to 1 month. Basal medium for reprogramming cellsReagentFinal concentrationAmountAscorbic Acid (500 μg/μL)50 μg/mL5 μLB27 Supplement (50×)1×1 mLGlutaMAX Supplement (100×)1×500 μLPen-Strep (10,000 U/mL)100 U/mL500 μLHEPES (1 M)25 mM1.25 mL2-Mercaptoethanol (55 mM)0.11 mM100 μLITS-X (100×)0.5×250 μLKnockOut Serum Replacement1%500 μLDMEM/F12N/A45.895 mLTotalN/A50 mLProtect from light and store at 4°C for up to 2 weeks. CRITICAL: 2-Mercaptoethanol is toxic if swallowed or inhaled. It causes serious eye damage and skin irritation. Wear personal protective equipment and use only under a chemical fume hood. Maintenance medium for reprogramming cellsReagentFinal concentrationAmountAscorbic acid (500 μg/μL)50 μg/mL5 μLGlutaMAX Supplement (100×)1×500 μLKnockOut Serum Replacement5%2.5 mLComplete Human Epithelial Cell Medium W/O FBSN/A46.995 mLTotalN/A50 mLProtect from light and store at 4°C for up to 2 weeks.
Step-by-step method details
Barcoding lentiviral Gateway destination vector
Timing: 4–5 days
This section aims to generate barcoded pLenti6/V5-DEST vectors.
- 1.Barcoding pLenti6/V5-DEST vector by PCR and gel extraction of PCR product:
- a.Prepare the PCR reaction master mix on ice as described in the table below.PCR reaction master mixReagentAmountCircular pLenti6/V5-DEST Vector10 ngQ5 High-Fidelity DNA Polymerase0.5 μL10 μM Barcode_Temp_Nhel_Forward Primer2.5 μL10 μM Barcode_Temp_Nhel_Reverse Primer2.5 μL5× Q5 Reaction Buffer10 μL10 mM dNTP Mix1 μLNuclease-Free Waterto 50 μLNote: 10× Multiome Gel Beads include a poly(dT) sequence that enables capture of 3′ poly-adenylated mRNA for gene expression (GEX) library. It cannot detect transcripts originating from exogeneous vectors with >1 kb distance between polyA and the ORF. This barcoding PCR reaction will introduce a 20-bp random oligo (N20) with NheI site into pLenti6/V5-DEST vector downstream of the ORF and 78 bp upstream of its 3′ LTR region, which contains the polyA. Inserting TF barcodes close to polyA enables their efficient capture and optimal library size for Illumina sequencing. One barcoding PCR reaction will generate a complex pool of barcoded vectors that can be used to barcode as many TFs as needed and the excess PCR product can be kept for future use. Designing individual barcode primers for barcoding PCR to avoid screening uniquely barcoded vectors is also feasible when working with less than three TFs.CRITICAL: Prepare 4–6 reactions to obtain enough PCR product after clean-up and to maintain its complexity.
- b.Gently mix the reaction by pipetting and centrifuge briefly. Start PCR using the cycling conditions described in the table below.PCR cycling conditionsStepsTemperatureTimeCyclesInitial Denaturation98°C30 s1Denaturation98°C10 s30 cyclesAnnealing and Extension72°C5 minFinal extension72°C10 min1Hold4°C∞
- c.Load PCR product on a 0.7% agarose gel and run gel at 120 V for 1 h. Expected PCR product, linear pLenti6/V5-DEST-Barcode vector with Nhel site at both ends, is 8,724 bp (Figure 1A). Gel-purify the PCR product following manufacturer’s instruction. Troubleshooting 1.Note: DNA fragments may exhibit a slight shift of towards higher molecular weight on agarose gel before clean-up (Figure 1A).Pause point: Store purified product at −20°C for long-term storage or proceed to the next step.Figure 1. Barcoding PCR product and morphological changes of cells during reprogramming(A) Gel image of barcoding PCR product showing the expected product at 8,724 bp.(B) Gel images of restriction digested barcoded vector pLenti6/V5-DEST vector-1 (left) and the in silico digestion using NEBcutter (right).(C) Cell morphological changes during reprogramming. Scale bar represents 100 μm.
- 2.Restriction digest of linear pLenti6/V5-DEST-Barcode vectors to create sticky ends, self-circularization and transformation:
- a.Prepare following reaction components and mix gently. Spin down quickly and incubate at 37°C for 1 h.Restriction digest mixReagentAmountLinear pLenti6/V5-DEST-Barcode Vector800 ngFastDigest Buffer (10×)2 μLNhel1 μLDpnl1 μLNuclease-Free Waterto 20 μLNote: Nhel creates sticky ends for self-circularization. Dpnl helps to remove circular template from barcoding PCR reaction in step 1.
- b.Gel-purify the restriction digested product following manufacturer’s instruction.
- c.Prepare following reaction components and mix thoroughly. Spin down quickly and incubate at 16°C for 12 − 20 h.DNA ligation mixReagentAmountRestriction Digested Linear pLenti6/V5-DEST-Barcode Vector50 ng10× T4 DNA Ligase Buffer5 μLT4 DNA Ligase1 μLNuclease-Free Waterto 50 μL
- d.Use up to 2 μL of the mixture for transformation of 50 μL ccdB Survival 2T1R competent cells. Troubleshooting 1.Pause point: Store ligated product at −20°C for up to 1 month or proceed to the next step.
- 3.Screening of pLenti6/V5-DEST vectors carrying unique barcodes:
- a.Screen single colonies and purify plasmids following manufacturer’s instruction.
- b.Verify pLenti6/V5-DEST-Barcode clones by restriction digest and obtain the barcode sequences of individual clones by Sanger sequencing.
- i.Prepare following reaction components and mix gently. Spin down quickly and incubate at 37°C for 1 h.Restriction digest mixReagentAmountpLenti6/V5-DEST-Barcode Vector500 ngFastDigest Buffer (10×)2 μLAflII1 μLXhol1 μLNhel1 μLNuclease-Free Waterto 20 μLNote: AflII and Xhol help to confirm that no rearrangement in the LTR regions of pLenti6/V5-DEST-Barcode vectors has taken place. Nhel helps to ensure the insertion of barcode.
- ii.Run restriction digested product on 0.7% agarose gel. Expected bands from correct clones are 3876 bp, 3656 bp, 960 bp and 220 bp (Figure 1B).
- iii.Obtain the barcode sequence for individual clones by Sanger sequencing using Sseq_Barcode primer (see key resources table). Troubleshooting 2.CRITICAL: Avoid using the barcodes that introduce new AflII, Xhol and Nhel sites.
Cloning of individual TFs into barcoded Gateway destination vectors
Timing: 4–5 days, depends on the number of TFs
This section is aimed at cloning individual TFs into barcoded pLenti6/V5-DEST vectors and preparing the plasmids of barcoded TFs for lentivirus production.
- 4.Gateway recombination cloning and plasmid purification:
- a.Clone the ORFs of individual TFs into barcoded pLenti6/V5-DEST vectors by LR reaction following manufacturer’s instruction.CRITICAL: Use different barcoded pLenti6/V5-DEST vectors for individual TFs to ensure each TF is labeled with a unique barcode.
- b.Use 1 μL of LR reaction for transformation of 50 μL Stbl3 competent cells.
- c.Screen for positive clones by restriction digest as described in step 3b and further verify the sequences of ORF and barcodes by Sanger sequencing using Sseq_ORF and Sseq_Barcode primers (see key resources table), respectively.
- d.Perform plasmid Midiprep for each barcoded factor following manufacturer’s instruction.
Reprogramming of pancreatic ductal-like cells from human fibroblasts
Timing: 3–6 weeks, depends on the desired time points for investigation
This section describes how to induce pancreatic ductal-like cells from human fibroblasts through lentiviral expression of defined TFs.
- 5.Lentivirus production in 293FT cells:Culture 293FT cells as per manufacturer’s instruction.CRITICAL: Follow biosafety level 2 (BSL2) guidelines while working with lentivirus and be careful with the storage and disposal of biohazard waste.
- a.Poly-L-lysine coating.
- i.Coat T75 flask with 3 mL of 0.001% poly-L-lysine in DPBS. Incubate at 20°C–25°C for 15−20 min with gentle rotation to ensure even distribution.
- ii.Wash twice with DPBS and once with sterile distilled water.
- iii.Air dry for at least 2 h.Note: Coat flasks under sterile hood and store air-dried flasks in sterile conditions at 20°C–25°C for up to 2 weeks.
- b.Day 0: Seed 293FT packaging cells in poly-L-lysine coated T75 flask in complete medium, at 5 × 10^6^ cells per flask.Note: For the maintenance of 293T cells, use complete medium containing 500 μg/mL Geneticin.
- c.Day 1: Change medium to 8 mL of serum reduced medium at 2 h prior to transfection and transfect cells with the combination of transgene plasmid, packaging plasmid psPAX2 and envelope plasmid pMD2.G as follow:
- i.Prepare two 5 mL tubes with following components for one T75 flask of cells and incubate at 20°C–25°C for 5 min.TubeReagentTube 148 μL Lipofectamine 2000 in 1 mL Opti-MEMTube 212 μg transgene plasmid + 9 μg psPAX2 + 3 μg pMD2.G in 1 mL Opti-MEM
- ii.Transfer the reagent in Tube 2 to Tube 1. Mix gently and incubate at 20°C–25°C for 15 min.
- iii.Add transfection mixture dropwise to the cell culture flask, gently swirl to evenly distribute the transfection reagents.
- d.Day 2: Change medium. Add 20 mL of fresh complete medium.Note: Medium change can be performed at 8–16 h after transfection. Long-time transfection will reduce cell viability as the cells are cultured in serum reduced medium at this stage.
- e.Day 4: Harvest virus:
- i.Collect 20 mL of cell supernatant and centrifuge at 500 × g for 10 min at 20°C–25°C.
- ii.Filter through 0.45 μm vacuum filters to remove cell debris.
- iii.Add 6.7 mL of Lenti-X concentrator to viral supernatant and mix gently by inverting tube. Incubate at 4°C for 2 h.
- iv.Centrifuge at 1,500 × g for 45 min at 4°C.
- v.Remove supernatant and gently resuspend the pellet in 100 μL of DMEM. Store at −80°C in aliquots.
- vi.Titer virus following manufacturer’s instruction. Troubleshooting 3.Note: Viral titers were determined using Perkin Elmer p24 ELISA Kit that represents physical titer based on the concentration of p24 protein: 1 pg/mL of p24 ≈ 10^4^ lentiviral particles/mL ≈ 100 TU/mL, when considering each lentiviral particle contains 2,000 molecules of p24. Of note, physical titer includes free p24 and defective viral particles in addition to the infectious viral particles. To be more accurate, we also performed the functional tittering (TU/mL) by transducing HFFs with pLenti6/V5-EGFP virus, followed by fluorescence tittering protocol for lentivirus from Addgene. For the same pLenti6/V5-EGFP virus preparation, the functional titer approximately corresponds to 1% of the physical titer from p24 ELISA assay. Based on this comparison, the physical titers of other lentiviruses were converted to functional titers for infecting HFFs by diving physical titers with 100. Then MOI was calculated as follows:
- 6.Matrigel coating:
- a.Thaw matrigel on ice and mix to homogeneity using pre-chilled pipets.CRITICAL: Matrigel will start to gel above 10°C. Keep matrigel on ice all the time while working. Pre-chill all plastics or media coming in contact with matrigel.
- b.Dilute matrigel in 1:200 using chilled DMEM and add 200 μL of diluted matrigel per well in 24-well plate. Incubate at 20°C–25°C for 1 h.Note: A dilution of 1:200 only results in a thin, non-gelled protein layer which mainly helps with cell attachment.
- c.Remove unbound material and rinse twice using DMEM.Pause point: If not using the plate immediately, add 250 μL of DMEM per well and keep the plate in cell culture incubator for up to 2 days. Do not dry the plate after matrigel coating.
- 7.Direct reprogramming of HFFs to pancreatic ductal-like cells:
- a.Day 0: Seed early-passage HFFs at 20,000 cells per well in matrigel-coated 24-well plate.Note: Scale the cell number based on the surface area if using other cell cultureware. Use the cells before passage 8.
- b.Day 1: Transduce with 6F pool with 8 μg/mL polybrene. Use multiplicity of infection (MOI) = 1 for SOX17, FOXA2 and PDX1; MOI = 2 for HNF1B, HNF6 and SOX9.Note: Although the cells are transduced with a pool of six TFs, cells carrying any sub-combinations of TFs from the 6F pool can also be present. This will result in the heterogeneity of reprogramming cells. Since ectopic TF-expressing cells can be identified based on their barcodes, the non-transduced cells without any barcodes can either be used as control cells or excluded from the analysis. Optionally, non-transduced cells can be eliminated during the experiment by a seven-day blasticidin selection at 5 μg/mL. The MOI used for each of the transduced TFs was determined based on their expression levels in ductal cells from adult human11 together with the earlier test results.1
- c.Day 2: Change to fresh fibroblast medium.
- d.Day 3: Change to basal medium for reprogramming cells supplemented with 100 ng/mL Activin A, 1 μM CHIR99021 and 50 ng/mL FGF7.
- e.Day 5: Change to basal medium for reprogramming cells supplemented with 100 ng/mL Activin A and 50 ng/mL FGF7.
- f.Day 7: Change to basal medium for reprogramming cells supplemented with 2 μM retinoic acid, 500 nM PD0325901 and 200 nM LDN193189.
- g.Day 8: Change to basal medium for reprogramming cells supplemented with 100 ng/mL Activin A and 500 nM PD0325901.
- h.From day 10 onwards, culture cells in maintenance medium for reprogramming cells. Pancreatic ductal-like cells with epithelial cell morphology start to appear at around day 21 (Figure 1C). Troubleshooting 4.Note: During cell reprogramming, passage cells at 1:2 ratio using Accutase when cells reach around 90% confluency and replate cells in fresh matrigel-coated plates.
Preparing library for FI-snMultiome-seq analysis
Timing: 3 days
This section describes library preparation for FI-snMultiome-seq.
- 8.Collect reprogramming cells at desired time points and isolate nuclei using demonstrated protocol CG000365 from 10× Genomics. Troubleshooting 5 and 6. CRITICAL: Using good quality cells is critical for the success of the experiments. Remove dead cells following the demonstrated protocol from 10× Genomics if cell viability is less than 90%. For balanced representation of different conditions in the analysis, isolate nuclei from individual conditions, count number of nuclei from each condition and pool the desired number of nuclei from different conditions for single-nuclei capture. Avoid pooling cells from different conditions before nuclei isolation.Note: The required number of nuclei to be profiled per condition depends on the experimental setting, e.g., the number of transduced TFs. For high quality (Grade A) nuclei, 800 nuclei per TF is a good starting point when overexpressing one TF. If the quality of nuclei is less optimal (Grade B), a minimum of 1,000 nuclei is recommend for a single-TF condition. Please refer to 10× Genomics instructions to assess the nuclei quality.
- 9.Prepare libraries for ATAC and GEX following user guide CG000338 from 10× Genomics.
- 10.Barcode library preparation for detecting TF barcodes:
- a.Prepare following reaction components on ice. Mix thoroughly and centrifuge briefly.PCR reaction master mixReagentAmountPre-amplified sample3 μLKAPA HiFi HotStart ReadyMix25 μL10 μM Truseq_Read2_Vec_Amp_Forward Primer2.5 μL10 μM Truseq_Read1_Amp_Reverse Primer2.5 μLNuclease-Free Waterto 50 μLNote: Pre-amplified sample is the purified product after step 4.3p from user guide CG000338.
- b.Start PCR using the cycling conditions described in the table below.PCR cycling conditionsStepsTemperatureTimeCyclesInitial denaturation95°C3 min1Denaturation98°C20 s22 cyclesAnnealing65°C15 sExtension72°C10 sFinal extension72°C1 min1Hold4°C∞Note: To ensure the libraries are minimally amplified, add SYBR Green I to PCR reaction master mix and run a qPCR test. Calculate the optimal cycle number for each sample by determining the number of cycles required to reach 1/3 of the maximum R.
- c.Cleanup with SPRIselect beads:
- i.Vortex the SPRIselect beads until fully resuspended. Add 40 μL (0.8×) SPRIselect beads to each sample. Pipette mix 10 times.
- ii.Incubate 5 min at 20°C–25°C.
- iii.Centrifuge briefly. Place on the 10× magnetic separator (magnet·High) until the solution clears.
- iv.Remove the supernatant.
- v.Add 200 μL 80% ethanol to the pellet. Wait 30 s. Remove the ethanol. Repeat it.
- vi.Centrifuge briefly. Place on the magnet·Low. Remove any remaining ethanol.
- vii.Remove the tube strip from the magnet. Immediately add 40.5 μL Buffer EB.
- viii.Pipette mix 10 times and incubate 2 min at 20°C–25°C.
- ix.Centrifuge briefly. Place on the magnet·Low until the solution clears.
- x.Transfer 40 μL sample to a new tube strip.Pause point: Store at −20°C for long-term storage or proceed to the next step.
- d.Prepare following reaction components on ice. Mix thoroughly and centrifuge briefly.PCR reaction master mixReagentAmountTemplate DNA from step 10.c50 ng2× KAPA HiFi HotStart ReadyMix25 μLDual Index TT Set A10 μLNuclease-Free Waterto 50 μLCRITICAL: Choose different indices for the samples in a multiplexed sequencing run.Note: Barcode libraries can be pooled in the same sequencing run for GEX libraries if the indices are compatible.
- e.Start PCR using the cycling conditions described in the table below.PCR cycling conditionsStepsTemperatureTimeCyclesInitial denaturation95°C3 min1Denaturation98°C20 s4 cyclesAnnealing65°C15 sExtension72°C10 sFinal extension72°C1 min1Hold4°C∞Note: To ensure the libraries are minimally amplified, add SYBR Green I to PCR reaction master mix and run a qPCR test. Calculate the optimal cycle number for each sample by determining the number of cycles required to reach 1/3 of the maximum R.
- f.Cleanup with SPRIselect beads:
- i.Vortex the SPRIselect beads until fully resuspended. Add 40 μL (0.8×) SPRIselect beads to each sample. Pipette mix 10 times.
- ii.Incubate 5 min at 20°C–25°C.
- iii.Centrifuge briefly. Place on the 10× magnetic separator (magnet·High) until the solution clears.
- iv.Remove the supernatant.
- v.Add 200 μL 80% ethanol to the pellet. Wait 30 s. Remove the ethanol. Repeat it.
- vi.Centrifuge briefly. Place on the magnet·Low. Remove any remaining ethanol.
- vii.Remove the tube strip from the magnet. Immediately add 30.5 μL Buffer EB.
- viii.Pipette mix and incubate 2 min at 20°C–25°C.
- ix.Centrifuge briefly. Place on the magnet·Low until the solution clears.
- x.Transfer 30 μL sample to a new tube strip.Pause point: Store the sample at −20°C for long term storage or proceed to the next step.
- g.Run 1 μL sample of ATAC, GEX and barcode libraries at 1:5 dilution on an Agilent TapeStation High Sensitivity D5000 ScreenTape to determine the average fragment size (Figures 2A–2C).Figure 2QC of final FI-snMultiome-seq librariesTapestation trace of barcode library (A), ATAC library (B) and GEX library (C).
- h.Perform library quantification for sequencer clustering using KAPA Library Quantification Kit following manufacturer’s instructions and determine the concentration based on fragment size derived from TapeStation trace.
- i.Sequence ATAC and GEX libraries following user guide CG000338. Sequence barcode library using the same sequencing parameters for GEX library at a minimum of 2,000 read pairs per nucleus.Optional: Determine fragment size using Bioanalyzer or LabChip.
Expected outcomes
The FI-snMultiome-seq protocol enables efficient capture of the TF expression from exogeneous expression vectors using a factor-indexing approach, which significantly improves the current 10× single cell multiome assay. This allows the identification and characterization of the cells that express all stochastic combinations of transduced TFs during direct reprogramming by reading the factor barcodes. Thus, it provides robust molecular analysis for comparing various reprogramming conditions and assessing the effect of individual TF in one experiment. Non-transduced cells can either be used as control or excluded from analysis to reduce the noise in the data. Notably, our methodology also enables segregating the expression of TFs from exogeneous vectors and endogenous genes by separately counting the transcripts from factor barcodes (exogeneous) and mRNA (endogenous). This allows the investigation of the effect of transduced TFs on its endogenous expression. Therefore, the application of FI-snMultiome-seq opens a path to study TF-mediated direct reprogramming at single-cell resolution, providing a comprehensive overview of the remodeling of transcriptomic and epigenomic landscape during transdifferentiation. Furthermore, the FI-snMultiome-seq protocol is versatile. The primers used for barcode insertion can easily be modified to work with other lentiviral constructs. Other than direct reprogramming, FI-snMultiome-seq can also be applied to other experimental conditions and cellular models that aim to study the cellular response to overexpression of genes from lentiviral constructs.
Quantification and statistical analysis
The data were processed and analyzed as described in Fei et al.1 TF barcodes were extracted from the fastq files of the barcode library using the Sequence Input/Output interface (SeqIO)4 from BioPython5 and raw sequencing data from GEX and ATAC libraries were processed with Cell Ranger ARC pipeline (v2.0.1) for demultiplexing, alignment, and feature counting, followed by subsequent analysis using Seurat (v4.1.1)6 and Signac (v1.7.09).7 The number of reads for each TF barcode were counted and added as a column to the metadata of the Seurat object. Cells with <1,000 RNA unique molecular identifier (UMI) counts or ATAC fragments, >100,000 RNA UMI counts or >500,000 ATAC fragments, and >30% mitochondrial RNA were excluded. For scRNA-seq, the raw UMI counts were normalized and scaled using SCTransform, regressing out cell cycle effect and the effect of the percentage of the UMI counts originating from mitochondrial genes. The top 3,000 variable genes were selected for principal-component analysis (PCA). For scATAC-seq, peaks were called using MACS2 (v2.2.7.1).8 Peaks on nonstandard chromosomes and GRCh38 blacklist regions were excluded, followed by frequency inverse document frequency (TF-IDF) normalization and latent semantic indexing (LSI) reduction. The 1–35 PCs from the scRNA-seq data and the 2–35 LSI dimensions from the scATAC-seq data were used for constructing the weighted-nearest neighbor (WNN) graph. Per-cell motif activity scores were calculated using the Signac RunChromVAR wrapper.9 Cell type scores were computed using ScType (v1.0)10 with markers for pancreatic cell types from the ScType database and fibroblast markers from literature.12
Limitations
FI-snMultiome-seq is based on lentiviral gene delivery. As with lentiviral expression systems, there could be potential silencing of viral expression after a few passages. This limits its application in analyzing mature reprogramming cells at late stages. Besides, cells carrying any sub-combinations from the TF pool can be present in theory when transducing a pool of TFs. However, it may happen that only some of the sub-combinations are present and the cell number of some conditions can be small. Thus, careful experimental planning and including additional cells for the sub-combination(s) of interest is important for optimal performance of FI-snMultiome-seq.
Troubleshooting
Problem 1
Low colony count after transformation (related to steps 1–2).
Potential solution
- •Confirm all the entry and destination clones by sequencing to check for the integrity of the BP and LR recombination sites.
- •Confirm ampicillin concentration.
- •Avoid multiple freeze-thaws of ligation product.
- •Repeat transformation using a new fresh batch of competent cells.
- •Purify the restriction digested product to remove contaminants and repeat the ligation.
- •Purify the barcoding PCR product to remove contaminants. Repeat the restriction digest and ligation.
Problem 2
Incorrect recombination of barcoded lentiviral vectors (related to step 3).
Potential solution
- •Always use Stbl3 competent cells or others that are suitable for propagation of lentiviral constructs.
- •Grow the bacterial culture at lower temperature such as 30°C.
- •Lower rotation speed to 200 rpm.
- •Use the dual combination of ampicillin and chloramphenicol for selection.
Problem 3
Low yield of lentiviruses (related to step 5).
Potential solution
- •Use early-passage 293FT cells.
- •Avoid using overconfluent cells for viral production.
- •Use fresh cell culture media.
- •Harvest viruses twice. Collect viral supernatant on Day 3, add 20 mL of fresh complete medium per flask and do a second collection on Day 4. Combine the viruses from two collections.
Problem 4
No morphological changes during reprogramming (related to step 7).
Potential solution
- •Use early-passage HFFs for reprogramming, no later than passage 8.
- •Confirm the recipes of reprogramming medium.
- •Avoid using expired supplements or medium.
- •Avoid exposing the cells to 20°C–25°C for too long while passaging cells and taking microscope images.
Problem 5
Low cell number of nuclei (related to step 8).
Potential solution
We recommend starting with more than 500,000 reprogramming cells for nuclei isolation.
Problem 6
Clumps and cell debris during nuclei isolation (related to step 8).
Potential solution
- •Wash cells with DPBS + 0.04% BSA for a total of 3 times before lysis to remove debris.
- •Optimize cell lysis to obtain nuclei in good quality.
- •Filter nuclei solution using 40 μm Flowmi Cell Strainer to remove large clumps.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Biswajyoti Sahu ([email protected]).
Technical contact
Technical questions on executing this protocol should be directed to and will be answered by the technical contact, Liangru Fei ([email protected]).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate new datasets. The code for extracting the TF barcode1 is available at GiHub: https://github.com/KaiyangZ/TF_barcode_extraction.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Fei L.Zhang K.Poddar N.Hautaniemi S.Sahu B.Single-cell epigenome analysis identifies molecular events controlling direct conversion of human fibroblasts to pancreatic ductal-like cells Dev. Cell 58202317011715.e 810.1016/j.devcel.2023.08.02337751683 · doi ↗ · pubmed ↗
- 2Rackham O.J.L.Firas J.Fang H.Oates M.E.Holmes M.L.Knaupp A.S.FANTOM Consortium Suzuki H.Nefzger C.M.Daub C.O.A predictive computational framework for direct reprogramming between human cell types Nat. Genet.48201633133510.1038/ng.348726780608 · doi ↗ · pubmed ↗
- 3Cahan P.Li H.Morris S.A.Lummertz da Rocha E.Daley G.Q.Collins J.J.Cell Net: Network Biology Applied to Stem Cell Engineering Cell 158201490391510.1016/j.cell.2014.07.02025126793 PMC 4233680 · doi ↗ · pubmed ↗
- 4Cock P.J.A.Antao T.Chang J.T.Chapman B.A.Cox C.J.Dalke A.Friedberg I.Hamelryck T.Kauff F.Wilczynski B.de Hoon M.J.L.Biopython: freely available Python tools for computational molecular biology and bioinformatics Bioinformatics 2520091422142310.1093/bioinformatics/btp 16319304878 PMC 2682512 · doi ↗ · pubmed ↗
- 5Roberts A.Chung H.W.Mishra G.Levskaya A.Bradbury J.Andor D.Narang S.Lester B.Gaffney C.Mohiuddin A.Scaling up models and data with t 5x and seqio J. Mach. Learn. Res.2420231810.48550/ar Xiv.2203.17189 · doi ↗
- 6Hao Y.Hao S.Andersen-Nissen E.Mauck W.M.Zheng S.Butler A.Lee M.J.Wilk A.J.Darby C.Zager M.Integrated analysis of multimodal single-cell data Cell 184202135733587.e 2910.1016/j.cell.2021.04.04834062119 PMC 8238499 · doi ↗ · pubmed ↗
- 7Stuart T.Srivastava A.Madad S.Lareau C.A.Satija R.Author Correction: Single-cell chromatin state analysis with Signac Nat. Methods 19202225710.1038/s 41592-022-01393-734997233 · doi ↗ · pubmed ↗
- 8Zhang Y.Liu T.Meyer C.A.Eeckhoute J.Johnson D.S.Bernstein B.E.Nusbaum C.Myers R.M.Brown M.Li W.Liu X.S.Model-based Analysis of Ch IP-Seq (MACS)Genome Biol.92008 R 13710.1186/gb-2008-9-9-r 13718798982 PMC 2592715 · doi ↗ · pubmed ↗