[Fe(µ2-OH)6]3− Linked Fe3O Triads: Mössbauer Evidence for Trigonal µ3-O2− or µ3-OH− Groups in Bridged versus Unbridged Complexes
D. Nirosha T. De Silva, Tyson N. Dais, Geoffrey B. Jameson, Casey G. Davies, Guy N. L. Jameson, Paul G. Plieger

TL;DR
This paper describes the synthesis and analysis of iron complexes with different bridging oxygen groups, using Mössbauer spectroscopy to confirm their structures.
Contribution
The study provides Mössbauer evidence distinguishing between µ3-OH and µ3-O bridged iron triads in complex structures.
Findings
Complex C1 contains µ3-OH bridged iron triads.
Complexes C2 and C3 contain µ3-O bridged iron triads.
Mössbauer spectroscopy confirms the bridging differences in the iron triads.
Abstract
The syntheses, coordination chemistry, and Mössbauer spectroscopy of hepta-iron(III) complexes using derivatised salicylaldoxime ligands from two categories; namely, ‘single-headed’ (H2L) and ‘double-headed’ (H4L) salicylaldoximes are described. All compounds presented here share a [Fe3-µ3-O] core in which the iron(III) ions are µ3-hydroxo-bridged in the complex C1 and µ3-oxo-bridged in C2 and C3. Each compound consists of 2 × [Fe3-µ3-O] triads that are linked via a central [Fe(µ2-OH)6]3− ion. In addition to the charge balance and microanalytical evidence, Mössbauer measurements support the fact that the triads in C1 are µ3-OH bridged and are µ3-O bridged in C2 and C3.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
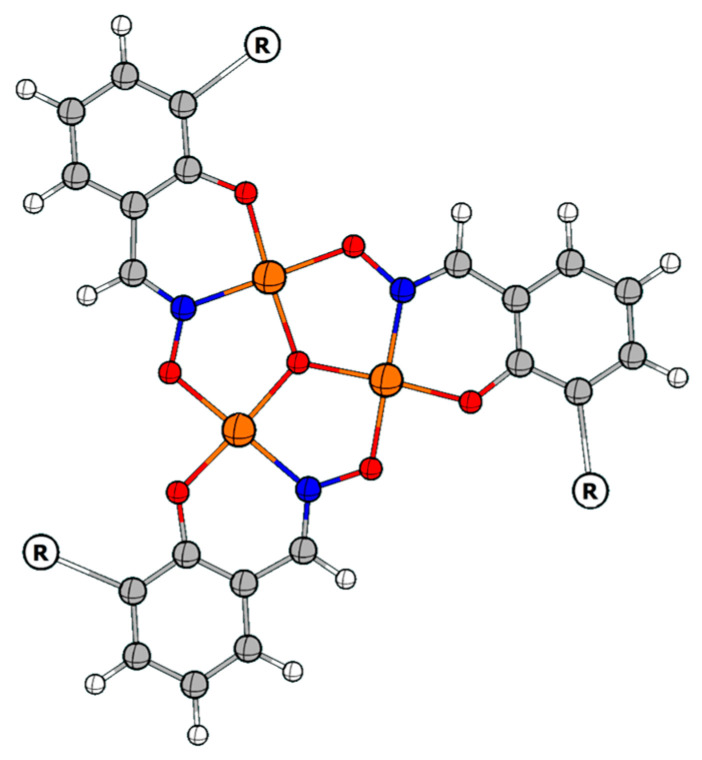 Figure 1
Figure 1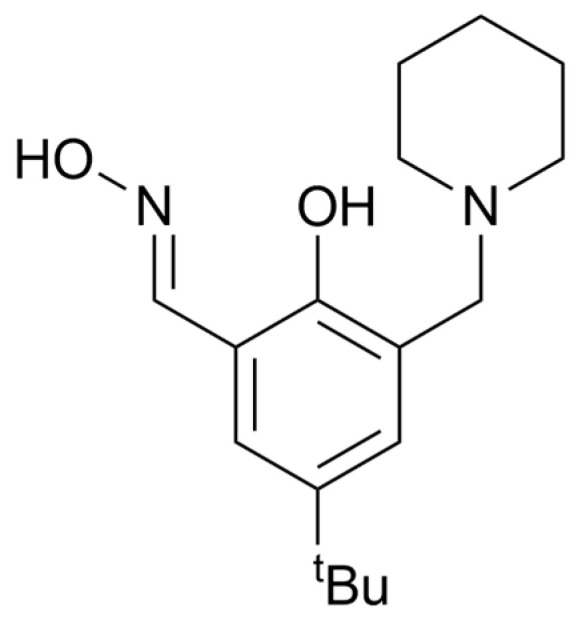 Figure 2
Figure 2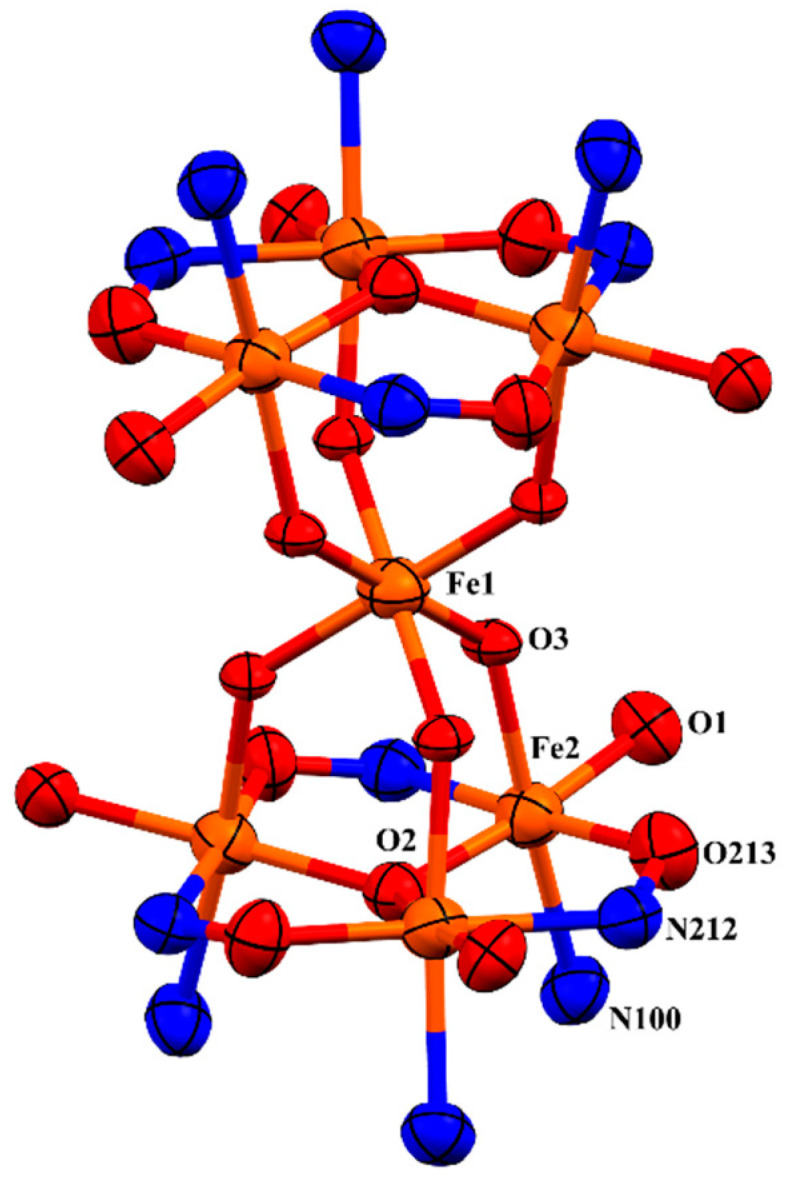 Figure 3
Figure 3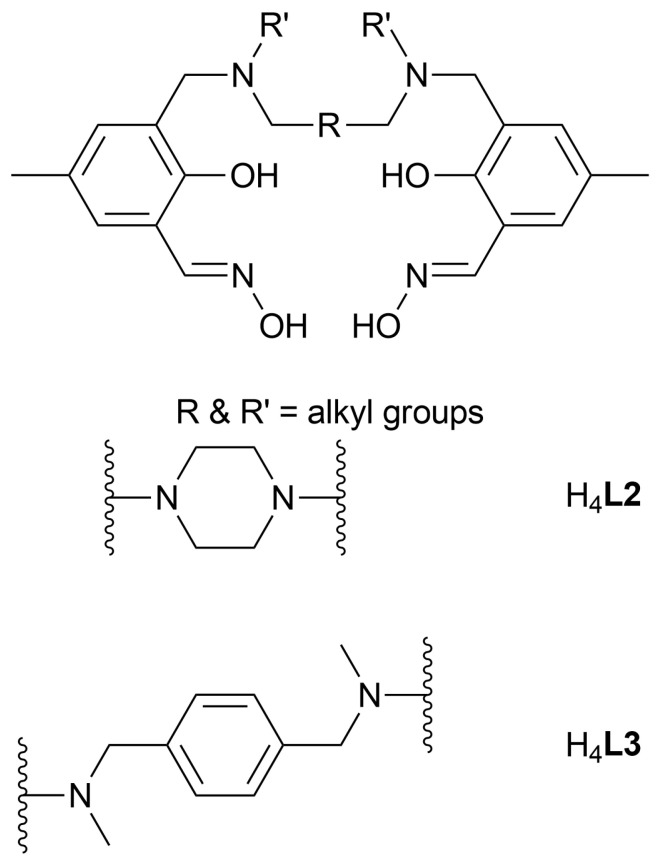 Figure 4
Figure 4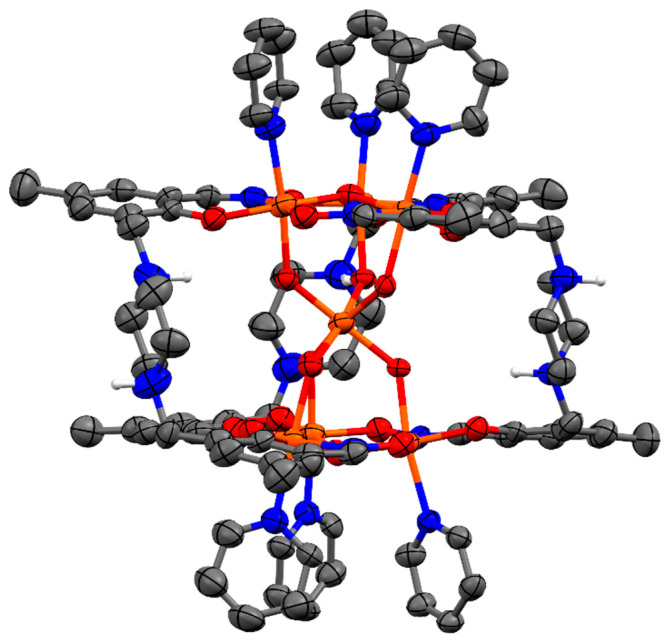 Figure 5
Figure 5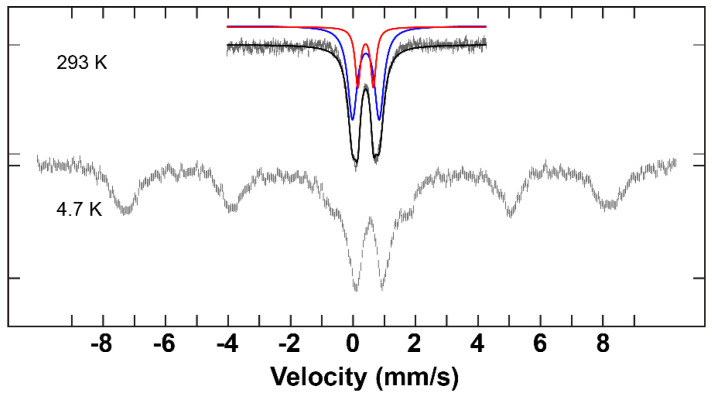 Figure 6
Figure 6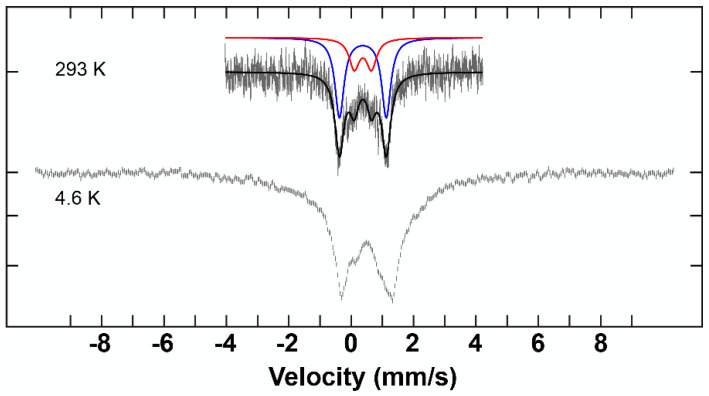 Figure 7
Figure 7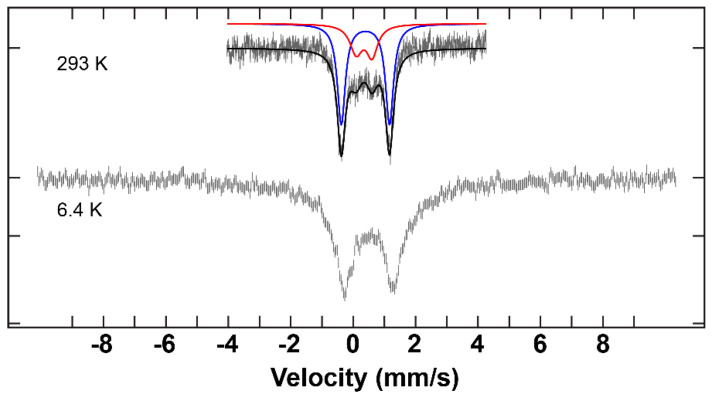 Figure 8
Figure 8Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMagnetism in coordination complexes · Metal complexes synthesis and properties · Metal-Catalyzed Oxygenation Mechanisms
1. Introduction
Polynuclear iron complexes have attracted interest due to their importance as both biological [1,2,3,4,5,6] and magnetic materials [7,8,9,10,11]. Herein, we report on the syntheses and coordination chemistry of three heptanuclear iron complexes built with derivatised salicylaldoximato ligands. The hepta-Fe(III) complexes presented here all share the common building block [Fe_3_O], in which Fe(III) ions are bridged by oximato- and oxo/hydroxo- groups. Salicylaldoximes and derivatised salicylaldoximes are well known to form multinuclear species that contain these triangular metal rings with three-fold symmetry (Figure 1) [12,13]. Study of this class of iron clusters has been fueled by the presence of analogous iron units observed in biologically important metalloproteins [2,14,15,16,17,18,19,20,21,22] and also as analogues of magnetically interesting manganese complexes [23]. The first iron–salicylaldoximato cluster reported was a tetra-iron species, [Fe_4_(saoH)4(sao)4] [24] of which the chemistry was later extended by Raptopolou et al. [13] to produce a tri-iron(III) compound with a [Fe_3_O]^7+^ core, coordinated by five benzoate ions and a salicylaldoximato di-anion.
A similar tri-iron(III) compound with the same core formed with six benzoate ions, an azido anion, and two bound ethanol molecules was reported by Boudalis et al. [12]. Recently, there have been several more examples reported for tri-, hexa-, and hepta-iron(III) salicylaldoximato/derivatised salicylaldoximato complexes containing the [Fe_3_O]^7+/8+^ core [23,25,26,27,28]. The first polynuclear copper complex with a linked derivatised salicylaldoximato ligand, N,N′-dimethyl-N,N′-hexamethylenebis(5*-tert*-butyl-2-hydroxy-3-hydroxyiminomethyl)benzylamine (H_4_L), was reported by Plieger et al. in 2009 [29]. This helical hexa-copper complex motivated us towards the synthesis of analogous iron(III) compounds with the same class of ligands [30]. However, in 2012, Brechin et al. [23] reported an iron(III) analogue of this hexa-copper complex using H_4_L [29]. This was the hepta-iron(III) cluster, [Fe_7_(µ_3_-O)2(µ_2_-OH)6(H_2_L-2H)3(pyr)6]·5BF_4_·6H_2_O·14MeOH, 1·2BF_4_·6H_2_O·14MeOH, consisting of two triangles of [Fe_3_O]^7+^, which are linked via a central [Fe(OH)6]^3−^ ion and three helical (H_4_L-2H) ligands.
Salicylaldoxime-based ligands are of particular interest due to the ease in derivatizing the aromatic ring and the inherent ability of the oximato moiety to coordinate multiple metal centres in close proximity. We herein report the syntheses and structures of three analogues (C1–C3) of the hepta-iron(III) complex 1 and use Mössbauer spectroscopy to evidence unexpected speciation of the central µ_3_-oxygen atom.
2. Results
2.1. Discussion of the Crystal Structure of the Fe7 Complex of a ‘Single-Headed’ Derivatised Salicylaldoxime (C1)
The first hepta-iron(III) compound, C1, was synthesised using a simple derivatised salicylaldoxime ligand, H_2_L1 (2-hydroxy-5-tert-butyl-3-(N-piperidinylmethyl)benzaldehyde oxime) (Figure 2) [31,32,33]. Slow evaporation of the filtrate from the 1:1 reaction mixture of Fe(BF_4_)2∙6H_2_O and the ligand, H_2_L1, in a methanol-pyridine solution led to the formation of dark maroon rhombic-shaped crystals of the hepta-iron(III) cluster, [Fe_7_(µ_3_-OH)2(µ_2_-OH)6(H_2_L1-2H)5(H_2_L1-H)(pyr)6]·(BF4)2·(H2O)6·(pyr)3 (C1·2BF_4_·8H_2_O·2pyr) which crystallised in the space group.
One sixth of the complex C1 represents the asymmetric unit, and the full complex is generated by an improper rotation. There are six molecules of the ligand H_2_L1 in the di-anionic form, H_2_L1-2H, in the complex, which are directly connected to six iron atoms (6 × µ_3_-Fe2) that form two metal triads of [Fe^III^3(μ_3_-OH)]^8+^, which are exactly parallel to each other (Figure 3). The central oxygen of the triad is formulated as a hydroxo species based on Mössbauer spectroscopy (see below). These triads are linked via six hydroxo groups that provide the coordination sphere to a seventh iron atom (Fe1), which sits in the middle of the complex as an anion [Fe(µ_2_-OH)6]^3−^ and is located 3.118 Å from the metal triads (the distance between the metal planes is 6.237 Å). Each triangle consists of three doubly deprotonated ligands (H_2_L1-2H), three iron(III) bound to a µ_3_-OH and three capping pyridine molecules (pyr). Thus, the positive charge (+21) provided by the seven Fe^III^ is overbalanced by −12 from the six ligands, −8 from hydroxo groups [2 × (µ_3_-OH) + 6 × (µ_2_-OH)], and −2 from 2 × BF_4_^−^ ions present within the lattice. Charge neutrality is achieved by a single proton distributed randomly over the 6 piperidinyl groups of the salicylaldoximato ligand.
Each iron atom of the complex is hexa-coordinated and sits in an approximately octahedral geometry. Equatorial sites around each iron atom of the triads (Fe2) are occupied by a phenolato oxygen (O1) atom and an oximato nitrogen (N212) atom from one ligand and an oximato oxygen (O213) atom from a neighbouring ligand and a central oxygen atom (µ_3_-O). A pyridine group (N100) and a hydroxo group (µ_2_-OH) are axially coordinated to each iron atom (Fe2) of the triangles. The iron centres of each metal triangle are held together by three N-O groups from the ligands resulting in a bridge between two neighbouring iron atoms. The bridging sequence is as Fe-O-N-Fe on both metal triangles. The central oxygen atom, µ_3_-O, of the metal triangle is displaced out of the metal planes by 0.314(6) Å away from the centre of the complex. The consequence is that the axial pyridyl groups tilt slightly away from each other, relieving steric strain. The Fe atom from [Fe(µ_2_-OH)6]^3−^ sits in an almost perfect octahedral coordination environment, as a consequence of sitting on the S_6_- axis. The hourglass-like metallic core of C1 is illustrated in Figure 3, and selected bond lengths and angles around Fe1 and Fe2 are shown in Table 1.
Additionally, water and pyridine molecules exist within the lattice. The hydroxo groups (µ_2_-OH) form strong hydrogen bonds (1.880 (10) Å) with water molecules and also moderately strong hydrogen bonds with neighbouring phenolate oxygen atoms, O1 (2.559 (6) Å) [34]. The composition of the crystal structure of this complex is confirmed by microanalytical data, charge balance, and Mössbauer.
2.2. Discussion of the Crystal Structures of Fe7 Complexes of Linked/’Double-Headed’ Derivatised Salicylaldoximes
The complexes, C2 and C3 are double-headed, µ_3_-oxo-bridged hepta-iron(III) compounds produced in the form of dark red rhombic crystals. Both were obtained by slow evaporation of filtered reaction mixtures of the iron salt Fe(BF_4_)2∙6H_2_O and the corresponding ligand (H_4_L2 and H_4_L3, respectively) in the presence of NaPF_6_ at a 1:2:2 ratio in a methanol-pyridine solution. These complexes are analogues of C1. Despite the different amine linkers present in the ligands (Figure 4) and the additional non-coordinated species present within the lattices, C2 and C3 are structurally very similar. Each of these clusters contains two approximately parallel oximato- and oxo-bridged metal triangles connected to a central Fe(III) atom via six hydroxo groups. X-ray crystal structures of the hepta-iron(III)clusters, C2 and C3, are described in this section. Selected structural parameters for these complexes can be found in Table 2.
Of particular note are the Fe-μ_3_-oxo bond lengths and displacements of the triply-bridging oxygen atom from the planes of the Fe_3_ moiety, which are not significantly different for the [Fe_3_^III^-µ_3_-OH]^8+^ moiety of C1, and the [Fe_3_^III^-µ_3_-O]^7+^ of 1 and C2 and C3. Therefore, there is no crystallographic evidence to distinguish µ_3_-O atoms being hydroxo in C1 from their being oxo in C2 and C3. In contrast to the [Fe_3_^III^-µ_3_-OH]^8+^ of C1, the metal triads are formulated as [Fe_3_^III^-µ_3_-O]^7+^ on the basis of Mössbauer spectroscopy. In C2, the oximato bridging sequence on the upper triangle is -N-O-, whereas it is -O-N- on the lower triangle. On the other hand, the same oximato bridging sequence occurs on both triangles of C3. As the ligands utilised for C2 and C3 are flexible linked salicylaldoximes containing salicylaldoxime units on either side, only three ligand molecules are required to form a hepta-iron(III)complex, unlike those used for C1. Three of these ‘salicylaldoxime heads’ from three ligand molecules form a lower triangle and the other three ‘heads’ form an upper triangle (Figure 1). Due to the flexibility of the di-amine linker between the salicylaldoxime ‘heads’, these complexes take a twisted helical shape (Figure 5).
2.3. Mössbauer Results and Discussion
^57^Fe Mössbauer measurements were performed on complexes C1–C3 at low and room temperature. Integral fits of the transmission were carried out for the data obtained at room temperature. The parameters for each of the samples are listed in Table 3.
The spectra that were recorded at 293 K illustrate two distinctive fitting lines (red and blue) (Figure 6, Figure 7 and Figure 8). These two lines can be unambiguously attributed to the two different iron environments present in each complex. The intensity of the blue peaks on the Mössbauer spectra of these complexes is much higher than that of the red peaks. The intensity ratio between the two iron species of each hepta-iron(III)compound was observed to be approximately 7:3, near enough to the expected value of 6:1 given by the crystallographic results, given that the central Fe atom is very tightly constrained relative to the iron triads. The isomer shift values of these complexes indicate the +3 oxidation state and high-spin state of the iron sites [35], and these numbers do not differ significantly among complexes C1–C3 at 293 K. The quadrupole splitting value for C1 (0.50 mm^−1^ and 0.87 mm^−1^), on the other hand, is significantly different from the values obtained for C2 and C3 (0.45–0.55 mm^−1^ and 1.50–1.55 mm^−1^) (see Table 3). The large quadrupole splitting (and relative intensity compared to the other doublet) is consistent with µ_3_-oxo groups for C2 and C3. The smaller quadrupole splitting of the doublets of weaker intensity for C2 and C3 and the pair of quadrupole doublets for C1 are consistent with µ_2_-hydroxo groups and for C1 the µ_3_-hydroxo groups.
3. Materials and Methods
All reactions were performed under aerobic conditions using chemicals and solvents as received, unless otherwise stated. ^1^H and ^13^C NMR spectra were recorded on a Bruker Avance 500 MHz spectrometer (Bruker, Billerica, MA, USA); δ values are relative to TMS or the corresponding solvent. Mass spectra were obtained using a Micromass ZMD 400 electrospray spectrometer (Waters Corporation, Millford, MS, USA). IR spectra were recorded on a Nicolet 5700 FT-IR spectrometer from Thermo Electron Corporation (Thermo Electron Scientific Instruments Corp., Madison, WI, USA) using an ATR sampling accessory. Elemental analyses were determined by the Campbell Microanalytical Laboratory at the University of Otago.
3.1. Synthesis of Ligands H4L2 and H4L3
The starting material of the multi-step ligand synthesis, 5-methylsalicylaldehyde, was synthesised as described in the literature [31]. The preparation of 3-(bromomethyl)-2-hydroxy-5-methylbenzaldehyde (1) and precursors L2a and L3a were carried out by the procedure of Tasker and Schröder [36]. The preparation of N,N′-dimethyl-p-xylenediamine (2), and the oximations were carried out according to the procedure by Plieger et al. [32] The ligand H_2_L1 was synthesised using the protocols in Tasker et al. [33] and Plieger et al. [37].
3.1.1. L2a (Precursor for H4L2): 3,3′-[1,4-Piperazinediylbis(methylene)]bis [2-hydroxy-5-methylbenzaldehyde]
Solutions of 1 (1.27 g, 5.54 mmol) and piperazine (0.242 g, 2.77 mmol), each in dry CH_2_Cl_2_ (15 mL), were simultaneously added to a stirred solution of Et_3_N (1.11 g, 11.0 mmol) in dry CH_2_Cl_2_ (20 mL). The resulting yellow solution was stirred for 24 h at room temperature (RT). The solution was washed with water (3 × 70 mL) and the organic phase dried over anhydrous Na_2_SO_4_. Removal of the solvent afforded a brown solid, which was purified by adding ethanol to a concentrated solution of the compound in CHCl_3_ affording a pale brown powder, which was dried in vacuo. Yield (0.90 g, 85%). MP 221–222 °C. υ_max_/cm^−1^ 1679 (s). Found: C, 68.14; H, 6.74; N, 7.26. Calc for C_22_H_26_N_2_O_4_·0.3C_2_H_5_OH: C, 68.44; H, 7.09; N, 7.04. ^1^H NMR (500 MHz; CDCl_3_) δ: 2.29 (s, 6H), 2.66 (br, 8H), 3.70 (s, 4H), 7.17 (d, J = 1.75 Hz, 2H), 7.41 (d, J = 1.61 Hz, 2H), 10.21 (s, 2H) ppm. ^13^C NMR (125 MHz; CDCl_3_) δ: 20.2, 52.4, 58.5, 122.0, 123.5, 128.4, 129.5, 137.0, 158.8, 192.6 ppm. m/z (ESI) 383 [M + H]^+^.
3.1.2. H4L2: 3,3′-[1,4-Piperazinediylbis(methylene)]bis [2-hydroxy-5-methylbenzaldehyde oxime]
A solution of hydroxylamine hydrochloride (0.400 g, 5.76 mmol) in dry ethanol (60 mL) was neutralised with potassium hydroxide (0.324 g, 5.76 mmol) in dry ethanol (60 mL). The resulting white precipitate was removed, and the filtrate was added to a solution of L2a (0.727 g, 1.90 mmol) in 5 mL chloroform and 95 mL dry ethanol over 30 min. The pale yellow solution was stirred for a further 24 h at RT, during which time a pale yellow precipitate was formed. The precipitate was filtered, and the remaining solvent was removed under reduced pressure. The combined pale yellow residues were then washed with cold chloroform (3 x 30 mL) and dried in vacuo. Yield (0.321 g, 41%). MP 245–246 °C. υ_max_/cm^−1^ 1625 (s), 1470 (s), 1136 (s), 822 (s). Found: C, 63.59; H, 6.84; N, 13.58. Calc for C_22_H_28_N_4_O_4_∙0.2C_2_H_5_OH: C, 63.80; H, 6.98; N, 13.29. ^1^H NMR (500 MHz; d6-DMSO) δ: 2.19 (s, 6H), 3.59 (s, 8H), 3.62 (s, 4H), 6.96 (d, J = 1.92 Hz, 2H), 7.23 (d, J = 1.74 Hz, 2H), 8.27 (s, 2H) ppm. ^13^C NMR (125 MHz; d_6_-DMSO) δ: 20.5, 52.4, 58.4, 118.5, 123.2, 126.5, 127.8, 131.6, 146.9, 153.6 ppm. m/z (ESI) 413 [M + H]^+^.
3.1.3. L3a (Precursor for H4L3): 3,3′-[1,4-Phenylenebis[methylene(methylimino)methylene]]bis [2-hydroxy-5-methylbenzaldehyde]
Solutions of 1 (1.27 g, 5.54 mmol) and 2 (0.461 g, 2.77 mmol), each in dry CH_2_Cl_2_ (15 mL), were simultaneously added to a stirred solution of Et_3_N (1.11 g, 11.0 mmol) in dry CH_2_Cl_2_ (20 mL). The yellow solution was stirred for 24 h at RT. The solution was washed with water (3 × 70 mL), and the organic phase dried over anhydrous Na_2_SO_4_. Removal of the solvent afforded a pale yellow solid, which was recrystallised by adding ethanol to a concentrated solution of the compound in CHCl_3_ affording yellow crystals, which were dried in vacuo. Yield (1.16 g, 90%). MP 152–155 °C. υ_max_/cm^−1^ 1678 (s), 3449 (br), 828 (s). Found: C, 72.60; H, 6.87; N, 6.03. Calc for C_28_H_32_N_2_O_4_: C, 73.02; H, 7.00; N, 6.08. ^1^H NMR (500 MHz; CDCl_3_) δ: 2.28 (s, 6H), 2.30 (s, 8H), 3.63 (s, 4H), 3.73 (s, 4H), 7.19 (d, J = 1.79 Hz, 2H), 7.34 (s, 4H), 7.44 (d, J = 1.57 Hz, 2H), 10.31 (s, 2H) ppm. ^13^C NMR (125 MHz; CDCl_3_) δ: 20.3, 41.5, 58.5, 58.5, 61.4, 122.3, 124.2, 128.3, 128.7, 129.4, 136.5, 136.6, 159.1, 192.1 ppm. m/z (ESI) 461 [M + H]^+^.
3.1.4. H4L3: 3,3′-[1,4-Phenylenebis[methylene(methylimino)methylene]]bis [2-hydroxy-5-methylbenzaldehyde oxime]
A solution of hydroxylamine hydrochloride (0.377 g, 5.43 mmol) in dry ethanol (60 mL) was neutralised with potassium hydroxide (0.323 g, 5.76 mmol) in dry ethanol (60 mL). The resulting white precipitate was removed, and the filtrate was added to a solution of L3a (1.00 g, 2.17 mmol) in 5 mL chloroform and 95 mL dry ethanol over 30 min. The pale yellow solution was stirred for a further 48 h at RT, after which time a pale yellow precipitate was obtained. The combined residues were filtered, washed with cold chloroform (3 × 30 mL) followed by cold ethanol (3 × 30 mL), and dried in vacuo. Yield (0.978 g, 92%). MP 203–204 °C. υ_max_/cm^−1^ 1610 (m), 2955 (m), 1469 (vs), 1285 (s), 1020 (m). Found: C, 67.01; H, 6.96; N, 10.89. Calc for C_28_H_34_N_4_O_4_·0.5C_2_H_5_OH: C, 67.81; H, 7.26; N, 10.91. ^1^H NMR (500 MHz; d_6_-DMSO) δ: 2.12 (s, 6H), 2.21 (s, 6H), 3.58 (s, 4H), 3.66 (s, 4H), 7.01 (d, J = 1.64 Hz, 2H), 7.24 (d, J = 1.80 Hz, 2H), 8.28 (s, 2H) ppm. ^13^C NMR (125 MHz; d_6_-DMSO): 20.5, 41.3, 58.1, 60.7, 118.5, 123.9, 126.3, 127.9, 129.5, 131.3, 137.1, 146.9, 153.6 ppm. m/z (ESI) 491 [M + H]^+^.
3.2. Synthesis of Metal Complexes C1–C3
3.2.1. [. Fe7(µ3-OH)2(µ2-OH)6(H2L1-2H)5(H2L1-H)1(pyr)6]·(BF4)2·(H2O)8 (pyr)2 (C1·2BF4·8H2O·2pyr)
To the ligand H_2_L1 (0.145 g, 0.50 mmol), dissolved in MeOH (12.5 mL), was added Fe(BF_4_)2∙6H_2_O (0.169 g, 0.50 mmol) in MeOH (12.5 mL). After full dissolution, NaPF_6_ (0.167 g, 1.00 mmol) and pyridine (2 mL) were added to the maroon-coloured solution. The mixture was stirred for 3 h and filtered, and the filtrate was left to evaporate slowly. X-ray quality crystals were produced after 2 weeks (CCDC 2331487). Yield (0.180 g, 67%). Found: C, 52.63; H, 6.35; N, 8.58. Calc for C_132_H_183_Fe_7_N_18_O_20_·2BF_4_·6H_2_O: C, 52.59; H, 6.52; N, 8.36. max/cm^−1^ 3388(br), 2967, 2370, 1605, 1550, 1459, 1084, 1040, 839, 732, 534, 437.
3.2.2. [Fe7O2(H4L2-2H)3(OH)6(pyr)6)]·(BF4)4·(H2O)7·PF6·(pyr)2 (C2·4BF4·7H2O·PF6·2pyr)
To the ligand H_4_L2 (0.206 g, 0.50 mmol), suspended in MeOH (12.5 mL), was added Fe(BF_4_)2∙6H_2_O (0.348 g, 1.00 mmol) dissolved in MeOH (12.5 mL). After full dissolution, NaPF_6_ (0.167 g, 1.00 mmol) and pyridine (2 mL) were added to the maroon-coloured solution. The solution was stirred for 3 h and filtered, and the filtrate was left to evaporate slowly. X-ray quality crystals were produced after 2 weeks (CCDC 2331488). Yield (0.200 g, 47%). Found: C, 40.24; H, 4.44; N, 8.84. Calc for C_96_H_114_Fe_7_N_18_O_20_·4BF_4_^−^·7H_2_O·PF_6_^−^: C, 40.47; H, 4.53; N, 8.85. υmax/cm^−1^ 3412(br), 1724, 1703, 1613, 1552, 1463, 1307, 1084, 1034, 825, 757, 483, 434.
3.2.3. [Fe7O2(H4L3-2H)2(H4L3-3H)(OH)6(pyr)6)]·(PF6)4·(H2O)7 (C3·4PF6·7H2O)
To the ligand H_4_L3 (0.245 g, 0.50 mmol), suspended in MeOH (12.5 mL), was added Fe(BF_4_)2∙6H_2_O (0.337 g, 1.00 mmol) dissolved in MeOH (12.5 mL). After full dissolution, NaPF_6_ (0.167 g, 1.00 mmol) and pyridine (2 mL) were added to the maroon-coloured solution. The mixture was stirred for 3 h and filtered, and the filtrate was left to evaporate slowly. X-ray quality crystals were produced after 2 weeks (CCDC 2331489). Found: C, 42.84; H, 4.24; N, 7.92. Calc for C_114_H_131_Fe_7_N_18_O_20_∙4PF_6_·7H_2_O: C, 43.19; H, 4.61; N, 7.95. υ_max_/cm^−1^ 3426(br), 2367, 1617, 1560, 1466, 1300, 1084, 1039, 823, 757, 618, 522, 440.
3.3. X-ray Structure Determination
X-ray data of complexes C1 and C2 were recorded at low temperature with a Rigaku-Spider X-ray diffractometer, comprising a Rigaku MM007 microfocus copper rotating-anode generator, high-flux Osmic monochromating and focusing multilayer mirror optics (Cu K_α_ radiation, λ = 1.54178 Å), and a curved image plate detector. CrystalClear [38] was utilized for data collection and FSProcess in PROCESS-AUTO [39] for cell refinement and data reduction.
Single-crystal diffraction data for C3 were collected at 100 K on the MX2 beamline (λ = 0.7093 Å) at the Australian Synchrotron, Victoria, Australia. The dataset was processed and evaluated using XDS [40]. The resulting reflections were scaled using AIMLESS140 from the CCP4 program suite [41]. All structures were solved employing direct methods and expanded by Fourier techniques [42]. All nonhydrogen atoms were refined using anisotropic thermal parameters. The hydrogen atoms were included in the ideal positions with fixed isotropic U value and were riding on their respective non-hydrogen atoms. Crystal data and refinement parameters for C1–C3 are given in Table A1. CCDC 2331487–2331489 contain the supplementary crystallographic data for this paper.
SQUEEZE results (electrons per formula unit):
C1: Electron count 126
C2: Electron count 232
C3: Electron count 276
3.4. Mössbauer Measurements
Samples of 17–29 mg were measured in a custom-made Teflon sample holder. Mössbauer spectra were recorded on a spectrometer from SEE Co. (Science Engineering & Education Co., Edina, MN, USA) equipped with a closed-cycle refrigerator system from Janis Research Co. and SHI (Sumitomo Heavy Industries Ltd., Shinagawa City, Tokyo, Japan). Data were collected in constant acceleration mode in transmission geometry. The zero velocity of the Mössbauer spectra refers to the centroid of the room temperature spectrum of a 25 µm metallic iron foil. Analysis of the spectra was conducted using the WMOSS program (SEE Co, formerly WEB Research Co., Edina, MN, USA).
4. Conclusions
The syntheses of one new ‘single-headed’ (C1) and two new ‘double-headed’ (C2 and C3) heptanuclear iron complexes formulated as [Fe_3_O–Fe(OH)6–Fe_3_O] are reported. Complexes C2 and C3 contain a common metallic core, [Fe_7_(µ_3_-O)2(µ_2_-OH)6]^+11^, which is structurally similar to the [Fe_7_(µ_3_-OH)2(µ_2_-OH)6]^+13^ core of C1. The presence of the µ_3_-OH groups within the iron triads of C1 is evidenced by ^57^Fe Mössbauer spectroscopy, observed as a significant change to the quadrupole splitting (0.50 mm^−1^ and 0.87 mm^−1^) which is consistent with the presence of µ_3_-OH groups.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Cohen I.A. Metal-metal interactions in metalloporphyrins, metalloproteins and metalloenzymes Struct. Bond.198040137
- 2Vincent J.B. Olivier-Lilley G.L. Averill B.A. Proteins containing oxo-bridged dinuclear iron centers: A bioinorganic perspective Chem. Rev.1990901447146710.1021/cr 00106 a 004 · doi ↗
- 3Horn A. Neves A. Bortoluzzi A.J. Drago V. Ortiz W.A. Crystal structure and magnetic properties of a new tetranuclear iron(III) complex with asymmetric iron coordination as a model for polynuclear iron proteins Inorg. Chem. Commun.2001417317610.1016/S 1387-7003(01)00156-3 · doi ↗
- 4Högbom M. Nordlund P. A protein carboxylate coordinated oxo-centered tri-nuclear iron complex with possible implications for ferritin mineralization FEBS Lett.200456717918210.1016/j.febslet.2004.04.06815178319 · doi ↗ · pubmed ↗
- 5Faiella M. Andreozzi C. Martin de Rosales R.T. Pavone V. Maglio O. Nastri F. De Grado W.F. Lombardi A. An artificial di-iron oxo-protein with phenol oxidase activity Nat. Chem. Biol.2009588288410.1038/nchembio.25719915535 PMC 3808167 · doi ↗ · pubmed ↗
- 6Nogueira M.L.C. Pastore A.J. Davidson V.L. Diversity of structures and functions of oxo-bridged non-heme diiron proteins Arch. Biochem. Biophys.20217051089173399149710.1016/j.abb.2021.108917 PMC 8165033 · doi ↗ · pubmed ↗
- 7Weighardt K. Pohl K. Jibril I. Huttner G. Hydrolysis products of the monomeric amine complex (C 6H 15N 3)Fe Cl 3: The structure of the octameric iron(III) cation of {[(C 6H 15N 3)6Fe 8(µ3-O)2(µ2-OH)12]Br 7(H 2O)}Br·8H 2O Angew. Chem. Int. Ed.198423777810.1002/anie.198400771 · doi ↗
- 8Barra A.L. Debrunner P. Gatteschi D. Schulz C.E. Sessoli R. Superparamagnetic-like behavior in an octanuclear iron cluster Europhys. Lett.19963513313810.1209/epl/i 1996-00544-3 · doi ↗