Patterns of Left Ventricular Remodelling in Children and Young Patients with Hypertrophic Cardiomyopathy
Emanuele Monda, Martina Caiazza, Chiara Cirillo, Marta Rubino, Federica Verrillo, Giuseppe Palmiero, Gaetano Diana, Annapaola Cirillo, Adelaide Fusco, Natale Guarnaccia, Pietro Buono, Giulia Frisso, Paolo Calabrò, Maria Giovanna Russo, Giuseppe Limongelli

TL;DR
This study explores how the heart's left ventricle changes over time in children with a specific heart condition called hypertrophic cardiomyopathy.
Contribution
The study identifies distinct patterns of heart remodelling in children with hypertrophic cardiomyopathy and highlights potential implications for early treatment.
Findings
Three distinct patterns of left ventricular remodelling were observed in children with hypertrophic cardiomyopathy.
Maximal left ventricular wall thickness was a predictor of hypokinetic end-stage heart disease during follow-up.
Some patients showed regression in wall thickness during childhood but experienced progression during adolescence.
Abstract
Introduction: The aim of this study was to evaluate the age at onset, clinical course, and patterns of left ventricular (LV) remodelling during follow-up in children and young patients with hypertrophic cardiomyopathy (HCM). Methods: We included consecutive patients with sarcomeric or non-syndromic HCM below 18 years old. Three pre-specified patterns of LV remodelling were assessed: maximal LV wall thickness (MLVWT) thickening; MLVWT thinning with preserved LV ejection fraction; and MLVWT thinning with progressive reduction in LV ejection fraction (hypokinetic end-stage evolution). Results: Fifty-three patients with sarcomeric/non-syndromic HCM (mean age 9.4 ± 5.5 years, 68% male) fulfilled the inclusion criteria. In total, 32 patients (60%) showed LV remodelling: 3 patients (6%) exhibited MLVWT thinning; 16 patients (30%) showed MLVWT thickening; and 13 patients (24%) progressed to…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
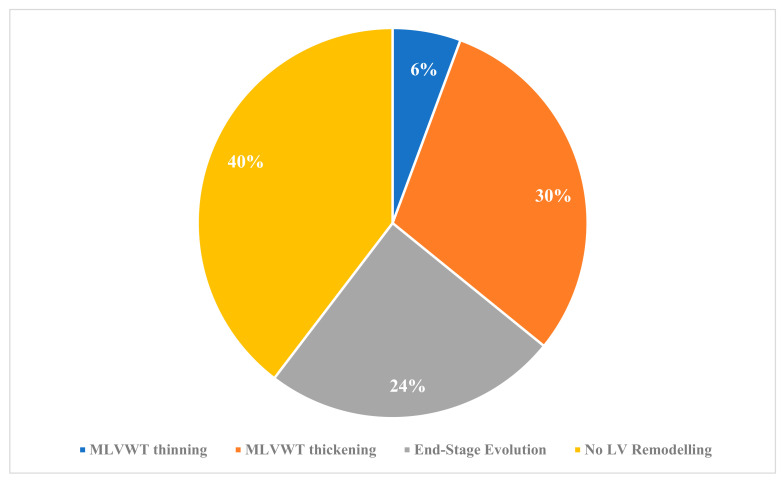 Figure 1
Figure 1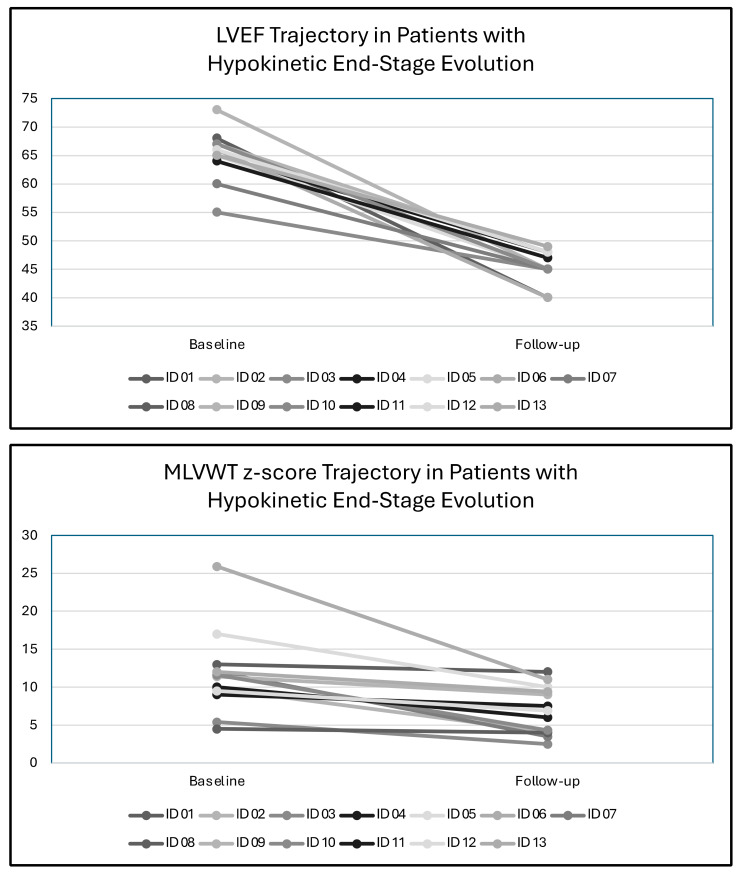 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiomyopathy and Myosin Studies · Cardiovascular Function and Risk Factors · Congenital Heart Disease Studies
1. Introduction
Hypertrophic cardiomyopathy (HCM) (OMIM #192600) is defined as a myocardial disease characterised by left ventricular (LV) hypertrophy not solely explained by hemodynamic overload [1,2]. Sarcomeric gene protein disease represents the most common cause of HCM, both in children and adults [1]. The identification of a sarcomeric gene pathogenic variant is observed in up to 60% of cases, with MYH7 and MYBPC3 representing the most commonly affected genes. However, in some cases (up to 5–10%), non-sarcomeric causes of HCM can be observed, including syndromic conditions, glycogen storage disorders, and mitochondrial and neuromuscular diseases [3]. The overall prevalence of HCM in children is estimated to be 0.002–0.005% [4].
Several myocardial diseases exhibit LV remodelling during follow-up, consisting of various changes in ventricular architecture, such as dilation of the LV cavity or myocardial thinning or thickening [5]. The pattern of LV remodelling has a significant impact on the disease course, potentially affecting the natural history and management of the condition.
While different LV remodelling patterns have been described in adults with sarcomeric HCM or in children with specific causes of HCM (e.g., Noonan syndrome with multiple lentigines, infants of diabetic mothers) [6,7,8,9], the clinical progression of LV hypertrophy in patients with sarcomeric or non-syndromic HCM during childhood or adolescence is poorly characterised. A better understanding of the natural disease course may help clinicians in improving risk stratification, predicting adverse outcomes, and personalising management, including the future timing of the introduction of disease-modifying therapies.
The aim of this study was to evaluate the age at onset, clinical course, and patterns of LV remodelling in children and young patients with sarcomeric or non-syndromic HCM.
2. Methods
A retrospective, longitudinal single-centre cohort of consecutive children (<18 years old) diagnosed with HCM between 2002 and 2018 was established. Clinical follow-ups were available up to December 2020. The study was conducted at a high-volume HCM centre: the Inherited and Rare Cardiovascular Diseases Unit, Department of Translational Medical Sciences, University of Campania “Luigi Vanvitelli”, Monaldi Hospital, Naples, Italy. Approval from the Internal Review Board Committee was obtained, and the study complies with the principles of Good Clinical Practice and the Declaration of Helsinki.
2.1. Eligibility Criteria
Patients diagnosed with sarcomeric or non-syndromic HCM under the age of 18 were eligible for inclusion. The diagnosis of HCM was confirmed if the LV wall thickness exceeded two standard deviations above the body surface area-corrected population mean (z-score ≥ 2), a finding not solely attributable to abnormal loading conditions [1,10]. Patients were classified as having sarcomeric HCM if they exhibited a pathogenic or likely pathogenic (P/LP) variant in one of the eight core sarcomeric genes: myosin-binding protein C (MYBPC3), myosin heavy chain (MYH7), cardiac troponin T (TNNT2), cardiac troponin I (TNNI3), a-tropomyosin (TPM1), myosin essential and regulatory light chains (MLY2, MYL3), and actin (ACTC). Patients were classified as having non-syndromic HCM if genetic testing did not identify a P/LP variant in a sarcomeric gene or if they did not undergo genetic testing, but other genetic syndromes, metabolic disorders, neuromuscular disorders, and congenital heart diseases (e.g., subaortic valve stenosis) were clinically excluded [11].
2.2. Data Collection
Eligible patients were identified by the principal investigator using multiple sources, including medical records and medical databases. The search strategy involved the use of keywords such as “hypertrophic cardiomyopathy” and “LV hypertrophy”. All cases diagnosed as HCM under the age of 18 were retrieved and then assessed for the inclusion and exclusion criteria. Anonymised clinical data were collected, encompassing non-identifiable demographics, family history, symptoms, resting ECG, 2D, Doppler, and colour transthoracic echocardiography, as well as genetic testing results. Clinical evaluations at our centre, including standard ECG and echocardiography, were conducted every 6 months. Data were collected from the baseline assessment to the last clinical review.
2.3. Clinical Investigations
Echocardiographic measurements were performed according to current guidelines [12]. Specifically, end-diastolic LV wall thickness was measured by 2D echocardiography in the parasternal short-axis view in four places at the level of the mitral valve and papillary muscles (anterior and posterior septum, lateral and posterior wall), and in two places at the apical level (anterior and posterior septum). Maximal LV wall thickness (MLVWT) was defined as the greatest thickness in any single segment [1].
2.4. Genetic Testing and Variant Classification
Patients underwent genetic analysis using a next-generation sequencing (NGS) panel containing 202 genes, including both sarcomeric and non-sarcomeric genes (e.g., RAS-MAPK genes, metabolic genes). Extensive details of the NGS panel and procedure have been previously described [13]. Genetic testing was performed after obtaining informed written consent.
2.5. Left Ventricular Remodelling
We defined a priori three different patterns of LV remodelling during follow-up: 1. an increase ≥15% in the MLVWT in both mm and z-score (MLVWT thickening); 2. a reduction ≥15% in the MLVWT in both mm and z-score (MLVWT thinning) not associated with a reduction in LV ejection fraction; and 3. a reduction of ≥15% in the MLVWT in both mm and z-score associated with a reduction in LV ejection fraction (hypokinetic end-stage evolution). A z-score that remained stable during follow-up was defined as having no LV remodelling or stability. The LV remodelling pattern was evaluated throughout the entire follow-up period and only in patients with ≥12 months of follow-up. We chose 15% as the cut-off to define MLVWT thinning or thickening in order to minimise potential bias related to inter- and intra-observer variability in echocardiographic LV measurements [5,14].
2.6. Statistical Analysis
Body surface area (BSA) was calculated from height and weight [15]. The 2D MLVWT is expressed in millimetres and z-scores corrected for BSA, using normative data validated in a large cohort of healthy individuals (http://www.parameterz.com/refs/lopez-circimaging-2017, accessed on 1 March 2024) [16]. MLVWT z-scores were recalculated retrospectively to give uniformity among patients evaluated in different eras. Normally distributed continuous variables are described as mean ± standard deviation, with two- or three-group comparisons conducted using Student’s t-test and analysis of variance (ANOVA), respectively. Skewed data are described as median (interquartile range [IQR]), with two- or three-group comparisons performed using Wilcoxon rank-sum and Kruskal–Wallis tests, respectively. Categorical variables are listed as numbers (percentage), with group comparisons conducted using a χ^2^ test or Fisher’s exact test. A significance level (p-value) of 0.05 (two-sided test) was used for all the comparisons. Univariate analysis of clinically relevant characteristics was performed. A stepwise regression, which included measures with a univariate p-value of ≤0.05, was used to build the multivariate model. Results are presented as odds ratios (ORs), 95% confidence intervals (CIs), and 2-sided p-values. All statistical analyses were performed using IBM SPSS Statistics for Macintosh, Version 27.0 (IBM Corp., Armonk, NY, USA).
3. Results
3.1. Enrolment and Baseline Characteristics
Among the 60 patients with a diagnosis of HCM at <18 years old, who were retrospectively identified, we excluded 7 patients with missing data at baseline evaluation or follow-up. The remaining 53 patients represent the final study cohort.
The clinical characteristics of the study population are shown in Table 1. The age at baseline evaluation was 8.8 ± 5.5 years, and 36 patients (68%) were male. A family history of HCM was present in 26 patients (49%), and a family history of sudden cardiac death (SCD) was present in 18 patients (34%). Among the 53 patients included in the study, 38 (72%) underwent genetic testing and 23 (44%) showed a P/LP variant in sarcomeric genes: 6 patients (11%) had a P/LP in MYBPC3, 12 (23%) in MYH7, 3 (6%) in TNNT2, and 2 (4%) in TPM1 (Table 2). ECG abnormalities were observed in 45 patients (85%). A review of the baseline echocardiograms demonstrated an average MLVWT of 16.9 ± 5.9 mm (z-score 9.1 ± 4.7). LV outflow tract obstruction was observed in 18 patients (33.9%) and required surgical myectomy in 3 cases.
3.2. Left Ventricular Remodelling
The different patterns of LV remodelling during follow-up are shown in Figure 1. Over a median follow-up of 9.4 ± 4.7 years, MLVWT thickening was the most common type of LV remodelling (n = 16, 30%), followed by hypokinetic end-stage evolution (n = 13, 24%) and MLVWT thinning (n = 3, 6%). No LV remodelling was observed in 21 patients (40%). Among these 21 patients, although there was no significant reduction in the absolute value of MLVWT, 10 patients exhibited a significant reduction in MLWVT z-score due to the increase in BSA during growth. In the three patients showing MLVWT thinning, the reduction in MLVWT was associated with an increase in LV end-diastolic diameter and no reduction in LVEF. In contrast, patients showing hypokinetic end-stage evolution showed a reduction in MLVWT and in LVEF (Figure 2).
3.3. Left Ventricular Remodelling According to Age at Presentation
Regarding diagnosis, 8 patients (15%) were diagnosed in infancy, 21 (39%) in childhood, and 24 (45%) in adolescence. Patients presenting in childhood or adolescence more commonly had a higher NYHA class and less commonly experienced MLVWT thinning during follow-up compared to those presenting in infancy. No significant differences in other clinical, genetic, and echocardiographic parameters were observed between the three groups (Table 3).
Two patients with sarcomeric HCM showed two different patterns of LV remodelling during follow-up. In the first patient, HCM was diagnosed at 1 month, with detection of MLVWT at the anterior septum equal to 12 mm (z-score 11.5). Septal thickness remained stable during follow-up (11.5 mm at 9 years old, z-score 2.8). However, at the last examination (16 years old), the patient showed an MLVWT of 26 mm (z-score 5.9). Patient 2 showed a similar course: HCM was diagnosed at 8 months (MLVWT 12 mm, z-score 5.6), and septal thickness remained stable during follow-up (12 mm at 14 years old, z-score 2.8) and showed progression during adolescence (18 mm at 17 years old, z-score +7.6).
3.4. Risk Factors for LV Remodelling during Follow-Up
Patients experiencing MLVWT thinning during follow-up were younger compared to those with other LV remodelling patterns or no LV remodelling. In contrast, patients with hypokinetic end-stage evolution had higher MLVWT at baseline evaluation than patients with other LV remodelling patterns or no remodelling (Table 4).
At univariate analysis, P/LP variants in MYH7 (OR 5.88 [95%CI 1.30–26.51], p-value 0.021), NYHA class II (OR 6.00 [95%CI 1.42–25.27], p-value 0.015), and MLVWT (OR 1.22 [95%CI 1.07–1.40] per 1 mm increase, p-value 0.003) were predictors of the hypokinetic end-stage remodelling pattern during follow-up. However, when accounting for MYH7 mutation status and NYHA class, the only risk factor maintaining its independent predictive value was MLVWT (OR 1.17 [95%CI 1.01–1.36] per 1 mm increase, p-value 0.043) (Table 5).
4. Discussion
HCM is a heterogeneous disease related to pathogenic variants in the sarcomeric gene in approximately 60% of patients [1,10,17]. Of clinical interest, the genetic background of HCM overlaps with other cardiomyopathies, such as restrictive and dilated cardiomyopathies [18].
Traditionally viewed as a disease of adolescents and adults, sarcomeric gene defects have also been reported in infants and children with HCM [19]. The natural history and clinical progression of LV hypertrophy have been extensively studied in adults, and the progression of LV hypertrophy in adults with HCM is the most common pattern of LV remodelling [5]. Additionally, the thinning of the myocardial wall associated with progressive systolic dysfunction, known as hypokinetic or end-stage HCM, is a well-documented phenomenon in a subgroup of patients [20], particularly in adults with unfavourable genotypes such as double or compound mutations, but it is less frequent in children, except in those forms associated with metabolic or mitochondrial disorders [19]. Recently, a favourable pattern of LV thinning, not associated with a reduction in LV ejection fraction and worse outcome, has been reported in adults with HCM [5]. However, the clinical course of LV hypertrophy in childhood HCM remains poorly characterised.
The main findings of this study were as follows: LV remodelling was observed in 60% of the patients, with MLVWT thickening representing the most common form; hypokinetic end-stage evolution was common, with MLVWT at baseline representing the most important risk factors at multivariate analysis; a specific pattern of remodelling was observed in two patients, with MLVWT regression during childhood and new progression during adolescence; and patients experiencing MLVWT thinning during follow-up were younger compared to patients presenting other LV remodelling patterns or no LV remodelling.
In our cohort of 53 patients with sarcomeric HCM, 30% showed a progression of MLVWT, 24% showed a hypokinetic end-stage evolution, and 6% showed LV thinning with preserved LV ejection fraction. These patterns have been already described in adults with HCM [5]. However, we observed a relatively high number of patients with end-stage evolution (24%). This is an important finding, since end-stage evolution is considered to be typically associated with mitochondrial and metabolic disorders [21]. This phenotype, particularly during adolescence, seems to be more frequent in patients with massive hypertrophy. Previous studies in young adults showed that HCM patients with massive LV hypertrophy had progressive LV wall thinning during follow-up, and this may predispose them to hypokinetic end-stage evolution, particularly in young patients [22,23,24]. This seems consistent with our finding, showing an association between massive LV hypertrophy and wall thinning/end-stage evolution. We also noted an apparently favourable evolution (LV wall thinning) in a small subgroup of patients with sarcomeric/non-syndromic HCM with diagnosis in infancy. Whether this is a real phenomenon or related to the lack of large normative data in this population is not known at this stage. In addition, long-term prospective observations are required to understand the clinical significance of these findings.
Moreover, in two patients with sarcomeric HCM, after a relative stabilisation during childhood, we noted a progression of LV hypertrophy during adolescence. This suggests another potentially important clinical implication of the study. It is intuitive that, after foetal life, adolescence represents another “hot phase” for cardiac remodelling. Patients with sarcomeric gene defects may be particularly sensitive to hormonal (e.g., IGF-1, GH) and environmental (e.g., physical activity) factors, which may trigger epigenetic modifications and plastic myocyte modifications [25]. In this sense, patients with an early clinical diagnosis of HCM and subsequent stabilisation/regression of LV hypertrophy during follow-up should be re-evaluated during adolescence, particularly if they carry a disease-causing sarcomeric gene pathogenic variant [25].
4.1. Clinical Implications
The results of our findings, along with experimental studies in the literature, indicate that the pathology of HCM may arise very early, even during cardiac development, and imply that the timing and target for future etiological therapies should be reconsidered [26]. Considering the potential impact of complex genotypes in different remodelling pattern evolution, an early clinical and genetic evaluation should be performed.
Sarcomeric mutations typically increase myofilament Ca^2+^ sensitivity and force generation in patients with HCM [27]. In mouse models with cardiomyopathies, the sarcomeric tension developed determines the type of remodelling (concentric in HCM versus eccentric in dilated cardiomyopathy) through differential down-stream activation of the mitogen-activated protein (MAP) kinase ERK1/2 [28]. Mavacamten, a small molecule inhibitor that reduces ATPase and force generation, is capable of blocking or reversing LV hypertrophy, fibrosis, and maladaptive genomic remodelling in an animal model of HCM and has been shown to improve LV outflow tract obstruction and clinical status in adults with HCM [29,30,31,32]. Hence, used at an early stage, it may be of potential therapeutic interest in children with HCM [33]. However, to date, there is a lack of data to support the safety and/or eligibility of this type of medication for the non-adult population.
4.2. Study Limitations
This study is limited by the small sample size and has limitations inherent to any retrospective analysis, which include missing data and survival bias. Moreover, genetic analysis was not performed for the entire cohort. Larger studies are needed to better characterise the clinical course of LV hypertrophy in infants and children with HCM and to better understand the possible clinical implications.
5. Conclusions
Different patterns of LV remodelling during growth were observed in a cohort of children with sarcomeric/non-syndromic HCM, including a relatively high number of HCM patients with hypokinetic end-stage evolution. Interestingly, a pattern of progressive MLVWT thinning/normalisation during childhood, with a new progression of MLVWT during adolescence, has been noted. A better knowledge of the remodelling mechanisms in children with sarcomeric HCM may be relevant to defining the timing and possible efficacy of new targeted therapies in the preclinical stage of the disease.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Arbelo E. Protonotarios A. Gimeno J.R. Arbustini E. Barriales-Villa R. Basso C. Bezzina C.R. Biagini E. Blom N.A. de Boer R.A. 2023 ESC Guidelines for the Management of Cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European So-ciety of Cardiology (ESC)Eur. Heart J.2023443503362610.1093/eurheartj/ehad 19437622657 · doi ↗ · pubmed ↗
- 2Lipshultz S.E. Law Y.M. Asante-Korang A. Austin E.D. Dipchand A.I. Everitt M.D. Hsu D.T. Lin K.Y. Price J.F. Wilkinson J.D. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement from the American Heart Association Circulation 2019140 e 9e 6810.1161/CIR.000000000000068231132865 · doi ↗ · pubmed ↗
- 3Maron B.J. Desai M.Y. Nishimura R.A. Spirito P. Rakowski H. Towbin J.A. Rowin E.J. Maron M.S. Sherrid M.V. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review J. Am. Coll. Cardiol.20227937238910.1016/j.jacc.2021.12.00235086660 · doi ↗ · pubmed ↗
- 4Mariani M.V. Pierucci N. Fanisio F. Laviola D. Silvetti G. Piro A. La Fazia V.M. Chimenti C. Rebecchi M. Drago F. Inherited Arrhythmias in the Pediatric Population: An Updated Overview Medicina 2024609410.3390/medicina 6001009438256355 PMC 10819657 · doi ↗ · pubmed ↗
- 5Musumeci M.B. Russo D. Limite L.R. Canepa M. Tini G. Casenghi M. Francia P. Adduci C. Pagannone E. MagrìD. Long-Term Left Ventricular Remodeling of Patients With Hypertrophic Cardiomyopathy Am. J. Cardiol.20181221924193110.1016/j.amjcard.2018.08.04130293658 · doi ↗ · pubmed ↗
- 6Monda E. Prosnitz A. Aiello R. Lioncino M. Norrish G. Caiazza M. Drago F. Beattie M. Tartaglia M. Russo M.G. Natural History of Hypertrophic Cardiomyopathy in Noonan Syndrome With Multiple Lentigines Circ. Genom. Precis. Med.20231635035810.1161/CIRCGEN.122.00386137199218 PMC 11791648 · doi ↗ · pubmed ↗
- 7Stegeman R. Paauw N.D. de Graaf R. van Loon R.L.E. Termote J.U.M. Breur J.M.P.J. The etiology of cardiac hypertrophy in infants Sci. Rep.2021191062610.1038/s 41598-021-90128-3PMC 813455634012105 · doi ↗ · pubmed ↗
- 8Rauen K.A. The RA Sopathies Annu. Rev. Genom. Hum. Genet.20131435536910.1146/annurev-genom-091212-153523 PMC 411567423875798 · doi ↗ · pubmed ↗