An mRNA vaccine for pancreatic cancer designed by applying in silico immunoinformatics and reverse vaccinology approaches
Md. Habib Ullah Masum, Shah Wajed, Md. Imam Hossain, Nusrat Rahman Moumi, Asma Talukder, Md. Mijanur Rahman

TL;DR
This paper describes the design of an mRNA vaccine for pancreatic cancer using computational methods to target immune receptors and enhance immune responses.
Contribution
The novel contribution is the in silico design of a pancreatic cancer mRNA vaccine targeting S100 family proteins with strong immune receptor binding and stability.
Findings
The vaccine showed high affinity for TLR-2 and TLR-4 receptors with binding energies of -141.07 and -271.72 kcal/mol.
The vaccine is predicted to be stable with a molecular weight of 165023.50 Da and a minimum free energy of -1760.00 kcal/mol.
The vaccine design induced memory B-cells and T-cells and increased helper T-cells and immunoglobulins.
Abstract
Pancreatic ductal adenocarcinoma is the most prevalent pancreatic cancer, which is considered a significant global health concern. Chemotherapy and surgery are the mainstays of current pancreatic cancer treatments; however, a few cases are suitable for surgery, and most of the cases will experience recurrent episodes. Compared to DNA or peptide vaccines, mRNA vaccines for pancreatic cancer have more promise because of their delivery, enhanced immune responses, and lower proneness to mutation. We constructed an mRNA vaccine by analyzing S100 family proteins, which are all major activators of receptors for advanced glycation end products. We applied immunoinformatic approaches, including physicochemical properties analysis, structural prediction and validation, molecular docking study, in silico cloning, and immune simulations. The designed mRNA vaccine was estimated to have a molecular…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
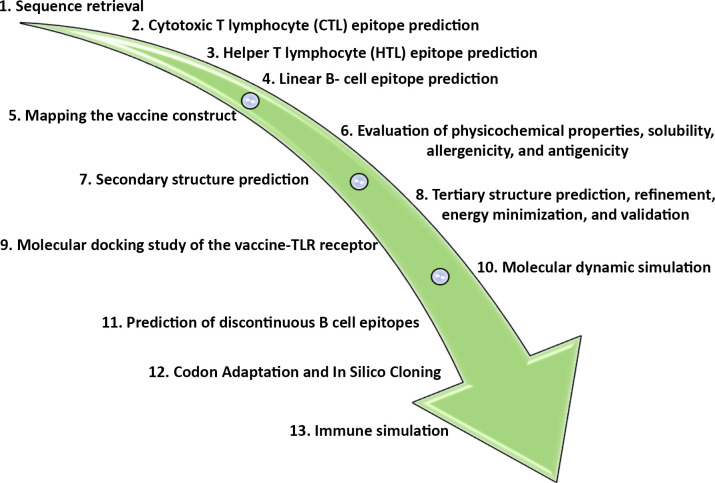 Fig 1
Fig 1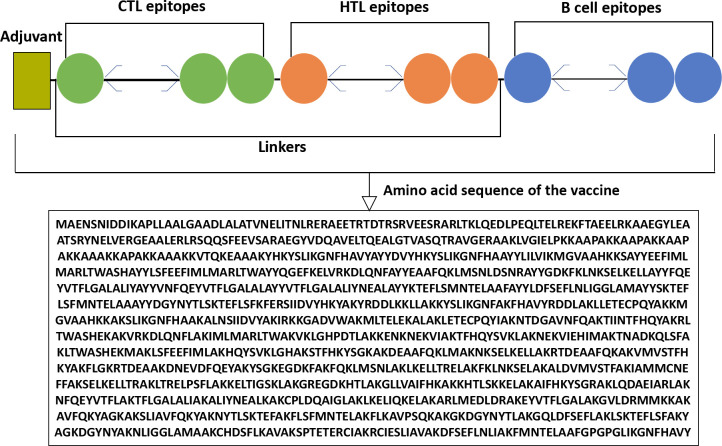 Fig 2
Fig 2 Fig 3
Fig 3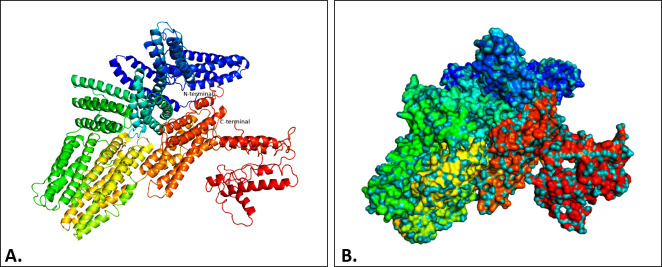 Fig 4
Fig 4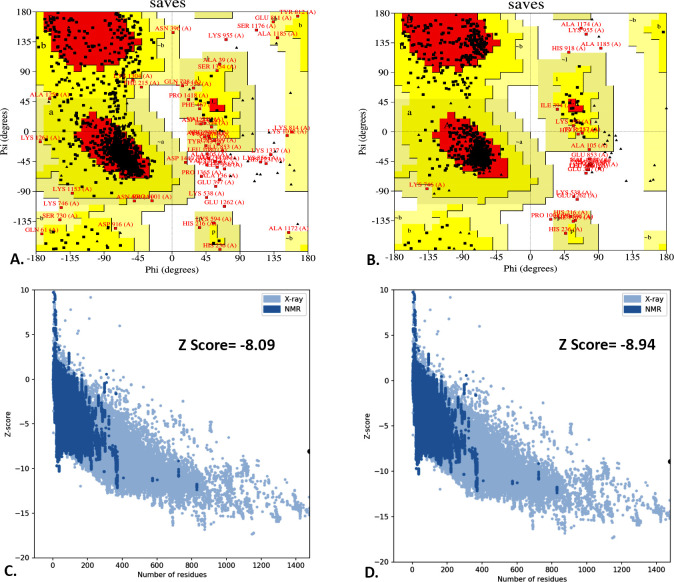 Fig 5
Fig 5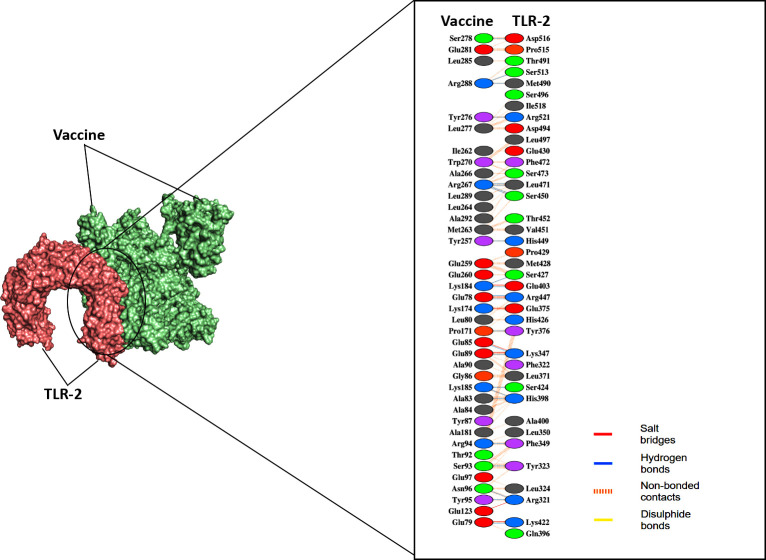 Fig 6
Fig 6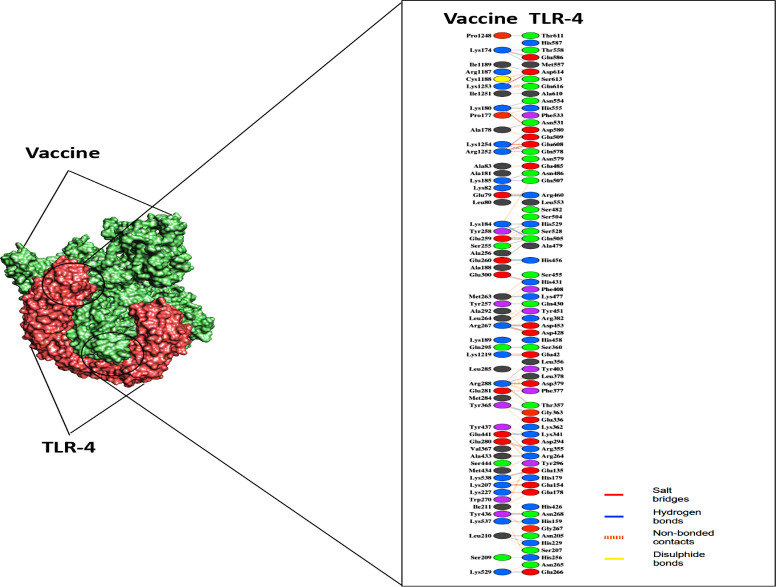 Fig 7
Fig 7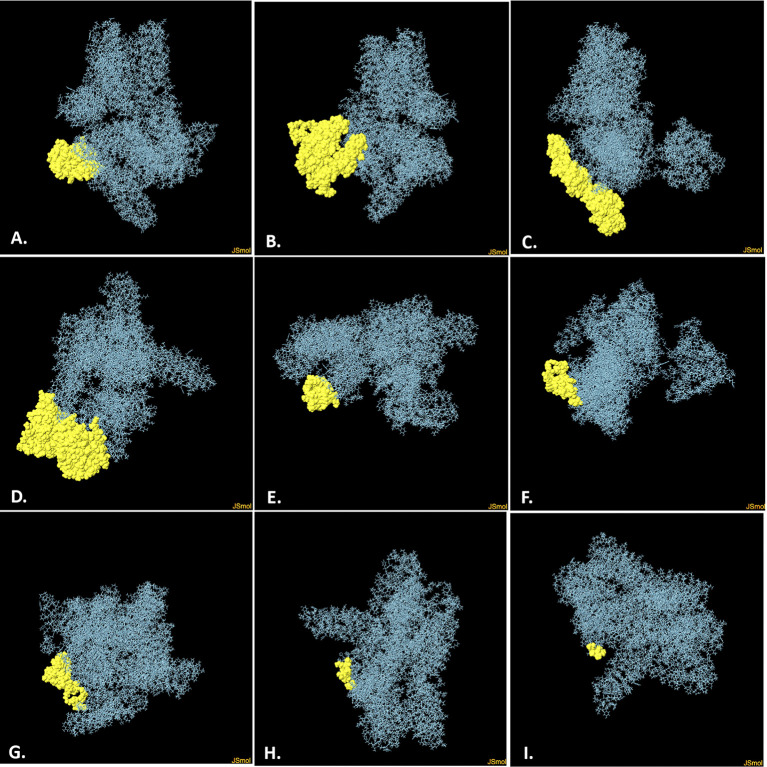 Fig 8
Fig 8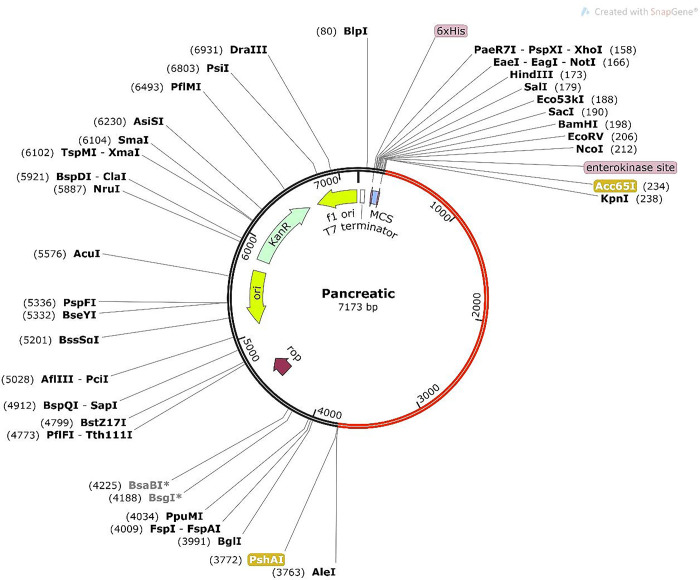 Fig 9
Fig 9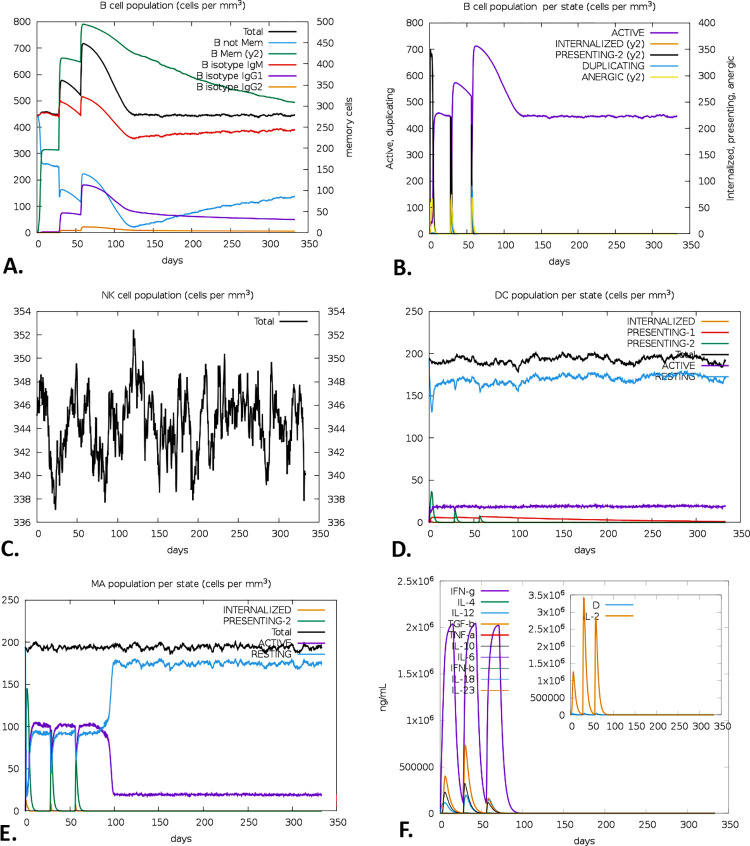 Fig 10
Fig 10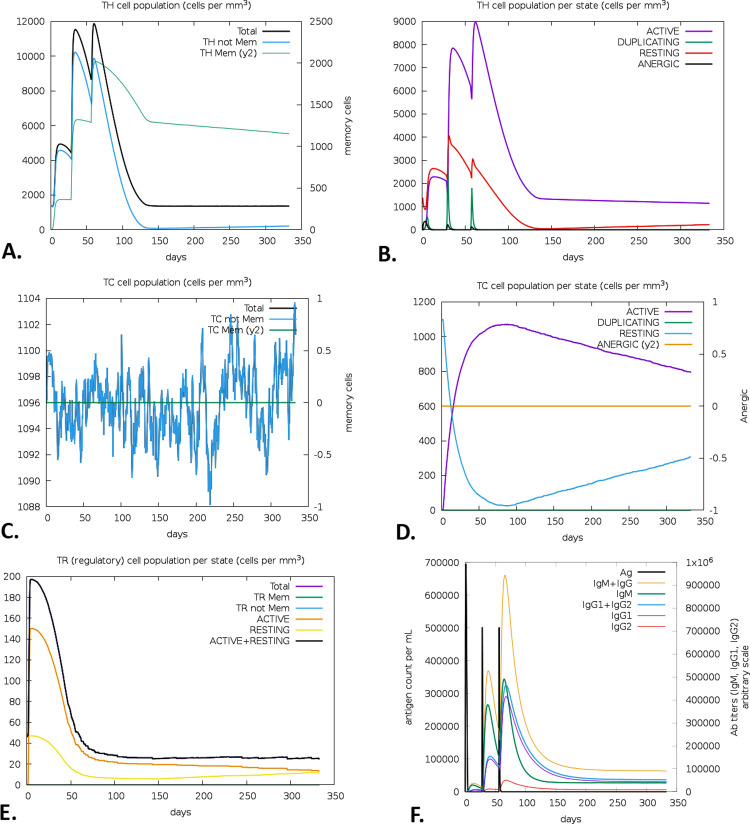 Fig 11
Fig 11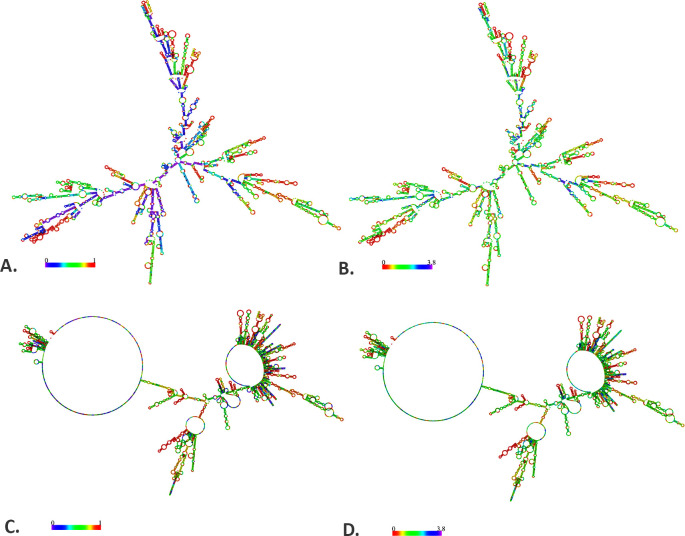 Fig 12
Fig 12Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Growth and Agriculture Techniques
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC), which constitutes 90% of pancreatic cancer (PC), is the fourth most prevalent cause of cancer-related mortality globally [1]. A study suggests that by 2030, the number of deaths in the US from PC will surpass that from breast, prostate, and colorectal cancer combined, partly as a result of improvements in the treatment of other cancers and an aging population [2]. In a recent study by the American Cancer Society (ACS) [3], the overall 5-year survival rate for PC is dramatically low, estimated at around 12%. The poor survival rate may be attributed to several factors, one of which is the late stage at which most patients are diagnosed [4]. Identifying the early stage of PC is challenging as there is a scarcity of symptoms and biomarkers that are precise to it [5]. In most cases, patients have already reached an incurable advanced stage by the time they exhibit symptoms and are diagnosed [5]. PDAC is primarily treated with chemotherapy and surgery, but because of distant metastasis at the diagnosis stage, the eligibility for surgical intervention is confined to a range of 15%-20% of patients [5]. Even when surgery is an alternative, approximately three-quarters of patients will have a recurrence within two years of surgery. Another developing approach for PC is neoadjuvant therapy, particularly for borderline and locally advanced unresectable cases [6]. Unfortunately, the randomized trial for neoadjuvant chemoradiotherapy in PC had to be stopped prematurely due to insufficient patient enrollment and outcomes that did not show statistical significance [7]. Based on different clinical and preclinical studies, mRNA-based therapeutics were found equal to or more effective than DNA or peptide platforms in delivering cancer vaccines [8]. The mRNA approach is adaptable and has effectively been employed in various vaccine delivery strategies, including systemic, subcutaneous, intramuscular, and in situ methods, as well as in genetically modifying dendritic cell-based vaccines and developing chimeric antigen receptor (CAR) T-cell therapies [8]. Also, mRNA-driven cancer vaccines encode complete cancer antigens, overcoming human leukocyte antigen restrictions for a wider immune reaction and remaining mutation-free due to mRNA’s inability to integrate into chromosomes [9].
Recent studies suggest that receptor for advanced glycation endproducts (RAGE) plays a significant role in the advancement of PC and might serve as a promising target for therapeutic interventions [10]. However, RAGE may be triggered by many members of the S100 protein family alongside being activated by other ligands [11]. S100 proteins are of notable importance in developing vaccines for PC, considering their unique attributes and implications for the progression of the disease [11, 12]. These proteins frequently appear in higher concentrations in PC tissues, serving as robust biomarkers for disease detection and prognosis. Their abnormal expression identifies malignant cells and contributes to their prospective targets for immunotherapeutic interventions [11, 12]. This offers the potential to develop vaccines that aim to stimulate an immune response against malignant tissues while excluding healthy ones. The S100 protein family consists of 21 members, which have a significant degree of structural similarity and regulate cellular responses by serving as both intracellular calcium (Ca^2+^) sensors and extracellular factors [13]. Among the different proteins, S100A4 is considered a risk factor for PC [14, 15], which does not express in normal tissues but is highly expressed in PC cells and related to the tumor-node-metastasis (TNM) staging and tumor size in PC [12]. S100A6 is a biomarker in PC lesions restricted to the nuclei in PC cells but not in the noncancerous tissues [16, 17]. S100A8 and S100A9 are two overexpressed proteins that are potential inflammatory mediators occurring in PDAC immunosuppression and suppress T-cell activation [18]. S100A11 is another potential gene therapy target, overexpressed in PC cells, and facilitates the PDAC interstitium and promotes PDAC growth [19].
In this study, we aimed to design a novel mRNA vaccine targeting five members of the S100 family protein, S100-A4, S100-A6, S100-A8, S100-A9, and S100-A11, which consists of cytotoxic T lymphocyte (CTL), helper T lymphocyte (HTL), linear B-cell epitopes derived from the selected proteins. With a combination of highly immunogenic adjuvants such as Heparin-binding hemagglutinin (HBHA) and five additional linkers, namely EAAAK, AYY, AK, KFER, and GPGPG, we designed the vaccine construct applying the immunoinformatic and computational strategies.
2. Methods
2.1 Retrieval of protein sequence
Amino acid sequences of the following proteins, S100-A4 (accession number: P26447.1), S100-A6 (accession number: P06703.1), S100-A8 (accession number: P05109.1), S100-A9 (accession number: P06702.1), S100-A11 (accession number: P31949.2) were retrieved from National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/) protein database and saved in FASTA format. The FASTA sequences were subsequently utilized for vaccine development. The summary of the study is depicted in Fig 1.
An overview of the study.
2.2 Cytotoxic T lymphocyte (CTL) epitope prediction
The Immune Epitope Database (IEDB) server (http://tools.iedb.org/mhci/) was applied to predict CTL epitopes from the intended protein sequences [20–30]. The server predicts CTL epitopes from a protein sequence based on affinity for major histocompatibility complex- I (MHC-I), the transportation efficiency (TAP), and the cleavage of the proteasome [21, 31]. We choose 12 MHC- I binding alleles, including HLA-A1, HLA-A2, HLA-A3, HLA-A24, HLA-A26, HLA-B7, HLA-B8, HLA-B27, HLA-B39, HLA-B44, HLA-B58, and HLA-B62 and applied the IEDB recommended method (NetMHCpan 4.1 EL) for this prediction. The predicted epitopes were further assessed for immunogenicity, antigenicity and toxicity through different validation tools like the IEDB class I immunogenicity (http://tools.iedb.org/immunogenicity/) [32, 33], Vaxijen v2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) [34] and ToxinPred Server (http://crdd.osdd.net/raghava/toxinpred/) [35, 36], respectively. The prediction threshold value was set to 0.5 for both Vaxijen v2.0 and ToxinPred server. However, the Vaxijen v2.0 has a prediction accuracy of 70% to 89%, while ToxinPred has a prediction accuracy of 94.50%.
2.3 Helper T lymphocyte (HTL) epitope prediction
The HTLs are recognized as having a variety of functions, including regulating T- and B-cells, identifying antigens through Major histocompatibility complex-II (MHC-II) on antigen-presenting cell (APC), helping in T-cell-mediated immunity, and so on [37]. Therefore, HTLs are crucial in developing adaptive immune responses [37]. Furthermore, the HTL epitope is critical for efficient vaccine development since vaccine antigen (Ag) is processed to be delivered via MHC-II [38, 39]. The IEDB MHC-II binding server was applied to screen HTL epitopes from intended protein sequences. The program was run by IEDB recommended method with 13 different MHC-II alleles, including HLA-DRB1-0101, HLA-DRB1-0301, HLA-DRB1-0401, HLA-DRB1-0701, HLA-DRB1-0801, HLA-DRB1-0901, HLA-DRB1-1001, HLA-DRB1-1101, HLA-DRB1-1201, HLA-DRB1-1301, HLA-DRB1-1401, HLA-DRB1-1501, and HLA-DRB1-1601. All the selected epitopes were further applied for the in silico assessment of interleukin-10 (IL-10) and interferon-gamma (IFN-γ) through IL-10Pred (http://crdd.osdd.net/raghava/IL-10pred/) and IFNepitope (http://crdd.osdd.net/raghava/ifnepitope/predict.php), respectively. However, the threshold value was set to -0.3 while predicting with IL-10pred, but it was set to 0.5 in the case of INFepitope. Additionally, the antigenicity and toxicity were also evaluated by Vaxijen v2.0 [34] and ToxinPred server [35, 36] with a threshold value of 0.5.
2.4 B-cell (linear) epitope prediction
The linear B-cell epitopes of the targeted proteins were predicted by the IEDB (http://tools.iedb.org/bcell/) and the Bepipred 2.0 (http://www.cbs.dtu.dk/services/BepiPred/) server [40]. To predict linear B-cell epitopes, the IEDB employs a combination of sequence features of the antigen, amino acid scales, Emini surface accessibility, and the Hidden Markov model (HMM) approach [41]. Meanwhile, the BepiPred 2.0 server utilizes an HMM and a propensity scale approach, which has a predicted accuracy of 73% [40]. In both instances, the threshold value was set as 0.5. Subsequently, the epitopes were assessed for allergenicity and antigenicity through the AllergenFP v.1.0 (http://ddg-ph armfac.net/AllergenFP/) [42] and VaxiJen 2.0 server (http://www.ddg-ph armfac.net/vaxijen/VaxiJen/VaxiJen.html), respectively [34].
2.5 Mapping the vaccine construct
All selected (CTL, HTL, and B-cell) epitopes from S100-A4, S100-A6, S100-A8, S100-A9, and S100-A11 proteins were utilized to construct the vaccine. The selected epitopes were linked together to develop a complete vaccine with recognized adjuvants and suitable linkers. Heparin-binding hemagglutinin (HBHA) (A5TZK3: HBHA_MYCTA) was added as an adjuvant, while five different types of linkers were used to connect the chosen epitopes: EAAAK, AYY, AK, KFER, and GPGPG [43, 44].
2.6 Evaluation of physicochemical properties, solubility, allergenicity, and antigenicity
The physicochemical characteristics of the vaccine were assessed following its construction by Expasy’s ProtParam (http://web.expasy.org/protparam/) server. These properties include the total amino acid count, composition and constitute of atoms, molecular weight and formula, total positive and negative residues, stability and aliphatic index, isoelectric point (pI), as well as extinction coefficients and grand average of hydropathicity (GRAVY) [45]. The allergenicity of the vaccine was evaluated by using the AllerTOP v. 2.0 server (https://www.ddg-pharmfac.net/AllerTOP/), which has a prediction accuracy of 85.3% [46]. Alongside, the antigenicity of the vaccine was also assessed by ANTIGENpro (http://scratch.proteomics.ics.uci.edu) (prediction accuracy of 82%) [47] and VaxiJen 2.0 (http://www.ddg-pharmfac.net/vaxijen/ VaxiJen/VaxiJen.html) (prediction accuracy of 70% to 89%) server [34]. In this analysis, the threshold value was set by default setting (0.5). Finally, the solubility of the vaccine was also evaluated by SOLpro (http://scratch.proteomics.ics.uci.edu) (prediction accuracy of 74%) [48–50] and Protein-Sol server (https://protein-sol.manchester.ac.uk/) (prediction accuracy of 58%) [50, 51].
2.7 Secondary structure prediction
The PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/), GOR4 (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_gor4.html) and SOPMA (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html) servers were applied to predict and assess the secondary structure of the vaccine [52]. With an accuracy of 84.2%, the PSIPRED predicts the secondary structure of a protein via neural network and PSI-BLAST (position-specific iterated BLAST) [53, 54]. The GOR4 server utilizes both information theory and Bayesian statistics, while the SOPMA uses a neural network to predict the secondary structure with an accuracy of 73.5% [55] and 69.5%, respectively [56]. The FASTA sequence was used to determine the secondary structure in the server mentioned above.
2.8 Tertiary structure prediction and validation
The I-TASSER server (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) predicted the vaccine’s tertiary structure (3D). The server utilizes various threading alignments and repeated template segment assembly simulations to determine a protein’s most accurate and precise tertiary structure [29, 44, 57, 58]. The server measures the structure’s confidence score (C-score) when assessing the quality of any predicted 3D model. Since an improved C-score signifies the highest quality or level of confidence of a predicted 3D model. Alongside, the template modeling score (TM-score) and root mean square deviation (RMSD) are typical measures of protein structure similarity, whereas subordinate values provide greater resolution and more accurate 3D model fits [44, 57, 59]. In terms of model prediction accuracy, the I-TASSER models may have an average error of 2 Å for RMSD and 0.08 for TM-score [60]. Consequently, the predicted 3D model of the vaccine was employed for structural refinement through the GalaxyWEB (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) server [61]. Further validation of the model was accomplished by the SAVES v6.0 server (https://saves.mbi.ucla.edu/). The server provides a Ramachandran plot, which defines the stereochemical quality of the predicted vaccine model [62–65]. To identify the structural accuracy of the predicted 3D model structure, we applied the ProSA-web server (https://prosa.services.came.sbg.ac.at/prosa.php). The server provides a Z-score for a predicted 3D model structure, which signifies the accuracy and the potential errors of the model structure [66, 67].
2.9 Molecular docking study of the vaccine-TLR receptor
The vaccine must effectively interact with the host’s immunological receptors to elicit a robust immune response. Therefore, protein-protein docking was used to predict the interaction of multi-epitope vaccines with immune receptors, toll-like receptor-2 (TLR-2) and TLR-4. The 3D structure of the vaccine and TLR-2 (PDB ID: 2Z7X) or TLR-4 (PDB ID: 3FXI) were applied to docking using the ClusPro 2.0 server (https://cluspro.bu.edu/login.php), which has a docking accuracy of ~71% [68–72]. However, both TLR-2 and TLR-4 have essential functions in vaccine-induced immunity [73]. These receptors can identify pathogen-associated molecular patterns (PAMPs) and initiate innate and adaptive immune responses. The TLR-2 mainly detects lipoproteins and lipopeptides, while the TLR-4 specifically detects lipopolysaccharides (LPS) [74]. Activating the TLR-2 and TLR-4 by vaccine components triggers a cascade of events that ultimately enhance adaptive immune responses. This includes improved antigen presentation, cytokine generation, and dendritic cell maturation. These processes, in turn, lead to increased antibody synthesis, T-cell activation, and the establishment of immunological memory, all of which significantly improve the effectiveness of vaccination [73, 74]. PyMOL (https://pymol.org/2/) and PDBsum (http://www.ebi.ac.uk/thornton-srv/databases/pdbsum/Generate.html) servers were used to analyze and visualize docked complex structures.
2.10 Free energy calculation by molecular mechanics with generalized Born and surface area solvation (MM-GBSA)
The free energy associated with the interaction between the "Vaccine—TLR-2" and "Vaccine—TLR-4" was calculated using MM-GBSA methodologies based on molecular mechanics and the Generalised Born approach. The molecular mechanics approaches under consideration include the influences stemming from bound interactions, van der Waals forces (VDW), electrostatic interactions (ELE), as well as polar (GB) and non-polar (SA) components [75–77]. The polar solvation component is calculated using the Generalised Born equation on the HawkDock server [75–77]. However, the accuracy of the MM-GBSA was reported to be 95.35% and 81.40% for the crystal and predicted structures, respectively [78].
2.11 Prediction of B-cell (discontinuous) epitopes
To predict the possible discontinuous B-cell epitopes of the vaccine, we applied the Ellipro of the IEDB database (http://tools.iedb.org/ellipro/) [79]. With the area under the ROC curve (AUC) value of 0.732 and prediction accuracy of 70%, the server utilizes three different algorithms to predict all possible discontinuous B-cell epitopes of the vaccine through their protrusion index (PI) values to illustrate an ellipsoidal protein shape and to quantify the residue PI and neighboring cluster residues [44, 79]. The selection parameters were set to default setting as a minimum score of 0.5 and a maximum distance of 6Å [79].
2.12 Codon adaptation and in silico cloning
The Java Codon Adaptation tool was employed to perform codon optimization of the vaccine for in silico cloning (http://www.jcat.de/) [80]. Therefore, we choose the Escherichia coli K12 strain as an expression vector for the vaccine. The codon adaptation index (CAI) value and GC content of the adapted sequence were also collected. Subsequently, the nucleotide sequence adapted to be compatible with the vaccine was introduced into the pET28a(+) vector through the restriction cloning module of the SnapGene software (https://www.snapgene.com/free-trial/). PshAI and Acc65I restriction sites were introduced to ensure suitable insertion into the plasmid.
2.13 Immune simulation
To perform the immune stimulation of the vaccine, the C-ImmSim server (https://kraken.iac.rm.cnr.it/C-IMMSIM/) was employed [81]. The server predicts the possible immune response of a mammalian immune system encountered by a vaccine injection. Both the humoral (antibody-mediated) and cellular (cell-mediated) responses were evaluated by this server [80, 82]. For the vaccine, a three-dose vaccination regime with a four-week interval was chosen. Nevertheless, the simulation parameters were configured with the default values, where the number of adjuvants and antigen injections were set to 100 and 1000, respectively [44]. Additionally, the time steps were defined as 1, 84, and 168, where each time step corresponds to 8 hours during daily life. Alongside, the simulation’s volume and steps were adjusted at 50 and 1000, respectively [44]. Without the interference of lipopolysaccharides (LPS), the random seed was set as 12345.
2.14 Structural validation of the mRNA vaccine
The secondary structure of the mRNA vaccine was predicted by the RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) web server [83]. The server can calculate the thermodynamically derived minimum free energy (MFE) of the query mRNA structures with an accuracy of 70% [84–86]. However, the energy parameters were set to default settings: a temperature of 37°C and a 1.021 molar (M) salt concentration. Upon acquiring the optimized DNA sequence via the JCat server, it was then transformed into a possible DNA sequence through the process of DNA<->RNA->Protein conversion at http://biomodel.uah.es/en/lab/cybertory/analysis/trans.htm. This was carried out to facilitate the analysis of mRNA folding and the secondary structure of the vaccine.
3. Result
3.1 Retrieval of protein sequence
The amino acid sequences of the proteins S100-A4, S100-A6, S100-A8, S100-A9, and S100-A11 were obtained from the NCBI protein database. These retrieved sequences were employed for further analysis.
3.2 Cytotoxic T lymphocyte (CTL) epitope prediction
CTL epitopes for five proteins were predicted through the IEDB web server based on their percentile rank (<1.0) and combined score (<1.0). Finally, a total of 73 epitopes were selected following the criteria: percentile rank and combined score <1.0, where 15 epitopes for S100-A4, 13 epitopes for S100-A6, 16 epitopes for protein S100-A8, 11 epitopes for protein S100-A9 and 18 epitopes for protein S100-A11. However, the epitopes were confined to 12 alleles (HLA-A1, HLA-A2, HLA-A3, HLA-A24, HLA-A26, HLA-B7, HLA-B8, HLA-B27, HLA-B39, HLA-B44, HLA-B58 and HLA-B62). The epitopes were assessed for immunogenicity, toxicity, and antigenicity, where they were found to be immunogenic, non-toxic, and antigenic (Table 1).
Table 1: The predicted CTL epitope with their immunogenicity, toxicity, and antigenicity.
3.3 Helper T lymphocyte (HTL) epitope prediction
HTL binding epitopes for the five proteins were predicted through the IEDB web server based on percentile rank <1.0 and screened out that can induce IFN-γ, IL-4, and IL-10 cytokines. Among the 15 selected HTL epitopes, seven were IL-4 non-inducers (non-positive value), and eight were negative to the production of IFN-γ (non-positive value). All of them were found to be IL-10 inducers with positive IL10 scores. Antigenicity, toxicity, and allergenicity were also assessed to select the predicted epitopes for the multi-epitope vaccine construction. We found that 15 epitopes showed antigenicity, non-toxic, and non-allergen activity (Table 2). However, the epitopes were confined to 13 alleles including HLA-DRB1-0101, HLA-DRB1-0301, HLA-DRB1-0401, HLA-DRB1-0701, HLA-DRB1-0801, HLA-DRB1-0901, HLA-DRB1-1001, HLA-DRB1-1101, HLA-DRB1-1201, HLA-DRB1-1301, HLA-DRB1-1401, HLA-DRB1-1501, and HLA-DRB1-1601.
Table 2: The predicted HTL epitopes of the selected proteins with their IFN-γ, IL-4, and IL-10 production capability, toxicity, antigenicity, and allergenicity.
3.4 B-cell (linear) epitope prediction
A total of 13 linear B-cell epitopes were chosen through the IEDB and the Bepipred 2.0 servers. Two peptide sequences from each protein, three from S100-A4, two from S100-A6, two from S100-A8, three from S100-A9, and three peptide sequences from S100-A11, were selected based on a bepipred score of > 0.5. The peptide sequences were evaluated for their allergenicity and antigenicity using the AllergenFP v.1.0 server and VaxiJen 2.0 server, respectively (Table 3). All of them were found to be probable non-allergens and antigens. However, the peptide sequences’ length varied from 8 to 35.
Table 3: The predicted B-cell (linear) epitopes.
3.5 Mapping the vaccine construct
From the selected proteins: S100-A4, S100-A6, S100-A8, S100-A9, and S100-A11, 73 CTL epitopes, 15 HTL epitopes, and 13 B-cell epitopes were chosen for the vaccine construct, which was built by connecting these epitopes with a suitable linker and a precise adjuvant. HBHA was tagged as an adjuvant to the N terminal, while the selected epitopes were linked to each other by using five linkers, including EAAAK, AYY, AK, KFER, and GPGPG (Fig 2).
The vaccine construct contains CTL (green color), HTL (orange color), and B-cell epitopes (blue color) with the linkers (adjacent line) and an adjuvant (olive color).
3.6 Evaluation of physicochemical properties, solubility, allergenicity, and antigenicity
The ExPASy ProtPram server illustrated that the vaccine has 1479 amino acids with an MW of 165023.50 Da. The vaccine had an isoelectric point (pI) of 9.45, indicating that it is basic (pH > 7) in nature. The vaccine also had 175 total number of negatively charged and 241 total number of positively charged residues. With the chemical formula of C_7496_H_11807_N_1983_O_2131_S_38,_ and a total number of atoms of 23455, the vaccine also had an extinction coefficient of 147770. However, the estimated high life of the vaccine was found to be different based on the expression system, while it was found to be 30 hours in mammalian reticulocytes, >20 hours in yeast cells, and >10 hours in E. coli. The server further confirmed the vaccine protein’s stability, which reported an instability index of 23.94 (instability index <40). The aliphatic index of the vaccine was found to be 79.02. Also, the vaccine is expected to be water-soluble (hydrophilic), with a GRAVY score of -0.440. The vaccine is predicted to be firmly soluble upon expression in E. coli (score of 0.999341). With estimated scores of 0.5283 and 0.4935, the Vaxijen 2.0 and ANTIGENpro servers suggested the vaccine had antigenic properties. The vaccine may not be responsible for any allergic reactions since no allergenicity was predicted in the vaccine through the AllerTOP v. 2.0 server (Table 4). However, the SOLpro and Protein-Sol servers predicted the vaccine as a soluble component with solubility scores of 0.999341 and 0.531, respectively.
Table 4: Physicochemical properties, solubility, allergenicity, and antigenicity of the vaccine construct.
3.7 Secondary structure prediction
The GOR4, SOPMA, and PSIPRED servers were employed to predict the secondary structure of the vaccine. The GOR4 server demonstrated that the vaccine’s structure comprised 70.18% alpha helix, 25.83% random coil, and 3.99% extended strands (beta sheet). Conversely, the SOPMA server’s prediction for the vaccine’s secondary structure revealed a random coil of 22.38%, an alpha helix of 66.13%, and an extended strand of 6.63%. The SOPMA server predicted a beta-turn structure of 4.87% in the vaccine; however, the GOR4 server did not detect any similar structure (Table 5). Finally, the PSIPRED server provided a three-state prediction for the protein secondary structure, including coil, helix, and strands (Fig 3, S1 Fig).
The secondary structure of the vaccine was predicted by the PSIPRED server.
Table 5: The vaccine’s secondary structure properties are predicted using GOR4 and SOPMA servers.
3.8 Tertiary structure prediction and validation
The I-TASSER server predicted five different models for the vaccine structure. Among these, the model was chosen to have the highest C-score of -0.05, TM-score of 0.71 ± 0.12, and RMSD score of 7.1 ± 4.2 Å (Fig 4). Subsequently, the refined 3D model was extracted from the GalaxyWEB server, which featured the RMSD, MolProbity score, and Ramachandran’s favorite region values of 0.404, 2.119, and 93.7%, respectively. The Ramachandran plot of the SAVES model demonstrated that the majority of amino acid residues (90.6%) were found in the most favored region, with a smaller percentage (7.5%) in the additional allowed region and a tiny percentage (0.8%) in the generously allowed region (Fig 5). In the unminimized model, the corresponding numbers were 81%, 15.1%, and 2.7%, respectively (Fig 5, Table 6). According to the ProSA server, the energy-minimized model exhibited a Z-score of -8.94 (Fig 5), while the unminimized model scored -8.09 (Fig 5). Additionally, the SWISS-MODEL predicts that the vaccine had a MolProbity score of 2.93, a Ramachandran preferred area of 81.31%, a QMEAN score of -7.88, and a QMEANDisCo Global score of 0.27± 0.05 before refinement, while after refinement these values were found to be 1.94, 94.11%, -3.88 and 0.29 ± 0.05, respectively (Table 6).
The predicted tertiary structure of the vaccine construct by I-TASSER.The ribbon (A) and surface (B) model view of the vaccine’s tertiary structure was visualized by PyMol software.
The Ramachandran plot and the Z-score of the predicted tertiary structure before (A and C) and after structural refinement (B and D).
Table 6: The quality assessment and structural validation of predicted tertiary structure.
3.9 Molecular docking between the vaccine and TLR receptor
The ClusPro 2.0 server was used to perform molecular docking and confirm the possible interactions of the construct with the TLR-2 and TLR-4 receptors. ClusPro 2.0 generated 60 docked structures for each receptor. Among these generated models, the preferred ones were chosen based on the highest binding affinity and the lowest intermolecular energy. When docking with TLR-2 and TLR-4, the predicted lowest energy scores were -1031.7 (kJ/mol) and -1313.6 (kJ/mol), respectively. Subsequently, PyMOL and PDB-sum were used to analyze and visualize the docked vaccine-TLRs complex structures, and based on the details provided by PDBsum, the "Vaccine—TLR-2" complex had seven hydrogen bonds, 24 salt bridges, and 251 non-bond interactions (Fig 6, S1 Table). Besides, the "Vaccine—TLR-4" complex contained 48 hydrogen bonds, 17 salt bridges, and 446 non-bond interactions (Fig 7, S1 Table).
The docked complex of “Vaccine—TLR-2” and their interacting amino acid residues predicted by the Cluspro 2.0 server.
The docked complex of “Vaccine—TLR-4” and their interacting amino acid residues predicted by the Cluspro 2.0 server.
3.10 Free energy calculation by MM-GBSA
We calculated the free binding energy (MM-GBSA) for the vaccine-receptors complexes through the HawkDock server. For the “Vaccine—TLR-2” complex, the VDW, ELE, GB, and SA were calculated to be -216.29 (kcal/mol), -1451.89 (kcal/mol), 1556.13 (kcal/mol), and -29.02 (kcal/mol), respectively. Subsequently, a total binding free energy of -141.07 (kcal/mol) was calculated for the complex. Regarding the “Vaccine—TLR-4” complex, the VDW, ELE, GB, and SA were estimated to be -356.85 (kcal/mol), -8958.65 (kcal/mol), 9092.05 (kcal/mol), and -48.28 (kcal/mol), respectively. However, an elevated binding free energy was calculated for the complex, estimating -271.72 (kcal/mol) (S2 Fig).
3.11 Prediction of B-cell (discontinuous) epitopes
With a total of 746 amino acid residues, the Ellipro server identified nine discontinuous B-cell epitopes in the vaccine (S1 Table). However, each of these epitopes has a number of residues and a score range ranging from 0.535 to 0.792. (Fig 8, S2 Table).
The predicted discontinuous B-cell epitopes of the vaccine (A–I).Yellow surfaces indicate the predicted discontinuous B-cell epitopes, while cyan sticks reveal the vaccine.
3.12 Codon adaptation and in silico cloning
In silico cloning was performed primarily to introduce the vaccine into the E. coli expression system. Following that, we adapted the codons of the vaccine in the E. coli K12 expression system through the JCAT server. In this analysis, we found that the GC content of the improved sequence was 47.4% while the CAI value was 1.0, which was satisfactory. The CAI is an approach for assessing the biases in synonymous codons for a certain target sequence. However, the CAI index’s value may vary from 0 to 1, suggesting the possibility of a successful expression. The higher the score, the more likely the target gene will be expressed. Meanwhile, the GC content is between 30 and 70%, corresponding to the optimal range. Finally, the optimized codon sequence was inserted in the pET28a (+) vector between PshAI and Acc65I restriction sites by Vector NTI Advance software (Fig 9).
In silico cloning of the vaccine’s optimized codon sequences into pET28a (+) vector.Restriction sites indicated in yellow boxes show the two restriction sites (Acc65I and PshAI).
3.13 Immune simulation
Within 60 days of receiving the immunization, a high expression of B-cells became apparent, along with an increased memory B-cell count (Fig 10A). Every B-cell was functioning, and the immune response persisted for almost a year (Fig 10B). The natural killer (NK) and dendritic cell (DC) cells also showed substantially long-lasting immunity (Fig 10C and 10D). After vaccination, the total macrophage (Mφ) population remained constant for a year (Fig 10E). Furthermore, the vaccination resulted in a substantial rise in the production of IFN-γ while concurrently suppressing the expression of tumor growth factor-β (TGF-β), conferring a robust immune response that persisted for two months (Fig 10F). Following vaccination, the observed T-cell responses include Th cells, CTL cells, and regulatory T-cells (Treg). The research further demonstrated a high expression level of both active Th cells and memory Th cells on day 60 following immunization, but that level declined with time (Fig 11A and 11B). Additionally, functional CTLs were identified as a high-level expression that remains relatively long (Fig 11C and 11D), whereas Treg cells reduced significantly after the immunization (Fig 11E). The total amount of antigens was observed for 50 days of vaccination, which was further replaced by IgM+IgG (Fig 11F).
Exploring the vaccine’s immune simulation using the C-ImmSim server.The evolution of entire (A) and per state (B) B-cell populations, NK (C) and DC (D) cell populations, the population of Mφ per state (E), and the cytokines and the IL-2 level are illustrated by the primary plot and the sub-plot, respectively (D) (Here, D refers to Simson’s index, which measures the degree of variety. Since an increase in D suggests an increase in the number of epitope-specific T-cells, a lower D value indicates a lower level of diversity).
T-cells mediated immune responses predicted by the C-ImmSim server.The evolution of Th with their memory cell life span (A), Th cell population per state cell (B), the development of entire Tc populations (C) and Tc population per state cell (D), the Treg populations per state (E), and the antigen and antibody titers after post vaccination state (F).
3.14 Structural validation of the mRNA vaccine
The secondary structure of the vaccine mRNA sequence was illustrated by the RNAfold server with an MFE score of -1760.00 kcal/mol (optimal secondary structure) and -1211.70 kcal/mol (centroid secondary structure). The free energy of the thermodynamic and the frequency of the MFE structure in the ensemble were predicted at -1818.19 kcal/mol and 0.00%, respectively. The prediction of the secondary structure of the mRNA vaccine is depicted in Fig 12 and S3 Fig. This result is consistent with previous research suggesting that the mRNA structure of the current vaccine may remain stable following its entry, transcription, and expression in the host [85, 87–90].
Predicted mRNA structure of the vaccine by RNAfold web server.The base pair probabilities of the mRNA vaccine with the minimum free energy (A) and centroid (B) structure and the positional entropy of the mRNA vaccine with the minimum free energy (C) and centroid (D) structure.
4. Discussion
In a notable scientific advancement, researchers successfully developed the first-ever cancer vaccine in the year 1980. This groundbreaking vaccine was created using tumor cells and tumor lysate, especially autologous tumor cells, in the development of colorectal cancer treatment [91, 92]. In the early 1990s, the discovery of the first human tumor antigen, melanoma-associated antigen 1 (MAGE-1), paved the way for further exploration and utilization of tumor antigens in developing potential cancer treatments [93]. Cancer vaccines primarily use tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs) to stimulate the individual’s immune system. In principle, the vaccine’s administration can elicit targeted cellular immunity and humoral immune response, impeding the progression of tumors and eventually eradicating malignant cells [94]. In the meantime, most cancer vaccines are undergoing preclinical and clinical trials [95]. Therefore, there is an urgency to develop more precise antigens and platforms for cancer vaccine development.
Over the last decade, significant advances in technology and research investment have shown that the fast-expanding area of mRNA therapeutic agents has become a viable platform for addressing many challenges encountered in vaccine development for infectious diseases and cancer [96, 97]. Since mRNA is a non-infectious and non-integrating platform, there are no possible hazards of infection or insertional mutagenesis, thus making it an advantageous vaccine candidate over subunit, killed, live-attenuated, and DNA-based vaccines [98–101]. Furthermore, the in vivo stability of mRNA may be modulated by several changes and delivery approaches since it undergoes degradation via intrinsic cellular mechanisms [98–101]. Additionally, there is also a prospect of modifying and downmodulating the immunogenicity of mRNA vaccines to improve their safety aspect [102]. mRNA vaccines may also have additional benefits, such as low production costs, rapid development, and vaccine effectiveness. Since they can be expressed in the cytoplasm without reaching into the nucleus, they perform better than DNA vaccines [103].
The manipulation of mRNA sequences has the potential to enable the manufacture of a wide range of targeted proteins with novel therapeutic applications. Computational approaches have emerged as more efficient for recognizing vaccine compositions than conventional vaccine development techniques. These approaches leverage the power of advanced algorithms and data analysis techniques to expedite the process of identifying optimal vaccine compositions. By analyzing vast amounts of data, these methods can quickly identify potential vaccine candidates and predict their efficacy [104]. Utilizing existing manufacturing processes offers advantages like enhanced diversity, flexibility, time efficiency, and cost-effectiveness in protein production [101, 105–107]. Despite being aware of some intrinsic constraints such as immunogenicity, instability, and delivery inefficiency, mRNA vaccines have shown promising signs owing to recent advancements in synthesis technology and structural alterations of mRNA sequences [108–110]. Recently, the FDA granted authorization over the first two SARS-CoV-2 mRNA vaccines, particularly Pfizer/BioNTech’s BNT162b2 and Moderna’s mRNA-127 (Spikevax) [111, 112]. Also, Pfizer/BioNTech’s BNT162b2 is the first mRNA vaccine to get commercial approval from the FDA [111, 112]. In 2023, Professors Katalin Kariko and Drew Weissman received a "Nobel prize" in physiology or medicine for the innovations of such SARS-CoV-2 mRNA vaccines [113]. The advent of these innovative vaccines has ushered in an age of innovation in the field of vaccination against infectious diseases and cancer.
The mRNA vaccine platform shows promise as a potential strategy for cancer vaccines since it involves the introduction of exogenous synthesized mRNA into cells to serve as templates for antigen production [97, 114]. Non-replicating mRNA and self-amplifying RNA vaccines are the two primary categories of mRNA vaccines. However, most mRNA-based cancer vaccines have been formulated by non-replicating mRNA [97, 103]. Moreover, mRNA vaccines enable the concurrent encoding of multiple antigens, including full-length tumor antigens. The elicitation of increased humoral and cellular immune responses by the encoded antigens enhances the potential to overcome resistance to cancer vaccines [115]. Also, the mRNA vaccine has shown promising results in stimulating MHC I-mediated CD8+ T-cell responses, which makes it a potential candidate for cancer treatment [116, 117]. Recently, Pfizer and BioNTech developed an mRNA neoantigen vaccine against PDAC. The vaccine is based on uridine mRNA-lipoplex nanoparticles, demonstrating a substantial efficacy level in the phase I clinical trial [118]. However, this study is the first endeavor to develop an in silico-based mRNA vaccine for pancreatic cancer. Therefore, this study involves the identification of various overexpressed protein members of the S100 protein family, including S100-A4, S100-A6, S100-A8, S100-A9, and S100-A11, to develop a successful multiepitope-based vaccine against pancreatic cancer. CTL epitopes are vital in stimulating the host immune responses to combat intracellular pathogens. The activation of Tc cell response originates through the binding of Tc cells to MHC-I molecules. Hence, vaccines formulated using CTL epitopes can induce robust CD8+ T cell activation, thus contributing a significant role in eradicating intracellular pathogens [119]. Furthermore, HTL epitopes are crucial in presenting immunogenic processed peptides to the T-cell receptor (TCR) on CD4+ T-cells. Therefore, it is pivotal in initiating both cellular and antibody-mediated immune responses. The association between MHC-II molecules and the TCR plays a key role in defense against microbial infections, rejecting transplants, and tracking the progression of malignancies [58, 120–123]. Consequently, the development of mRNA vaccines always requires recognition by CD4+ and CD8+ T-cells [80, 124, 125]. This study evaluated the epitopes from S100-A4, S100-A6, S100-A8, S100-A9, and S100-A11 for their ability to bind to MHC-I and MHC-II on immune cells. Regarding CTL epitopes, the presentation of peptides tends to be restricted to specific alleles, including HLA-A1, HLA-A2, HLA-A3, HLA-A24, HLA-A26, HLA-B7, HLA-B8, HLA-B27, HLA-B39, HLA-B44, HLA-B58 and HLA-B62. The HTL-predicted peptides were restricted to specific alleles, including MHC-II alleles such as HLA-DRB1-0101, HLA-DRB1-0301, HLA-DRB1-0401, HLA-DRB1-0701, HLA-DRB1-0801, HLA-DRB1-0901, HLA-DRB1-1001, HLA-DRB1-1101, HLA-DRB1-1201, HLA-DRB1-1301, HLA-DRB1-1401, HLA-DRB1-1501, and HLA-DRB1-1601. The peptides demonstrated an elevated level of antigenicity while showing minimal levels of allergenicity and toxicity.
B-cell epitopes have been extensively recognized as a fundamental aspect in the development of vaccines since they play a substantial role in the association between antigens and antibodies [80, 126–128]. The B-cell epitopes we anticipated exhibited an elevated antigenicity level and lacked any allergenicity indications. Subsequently, various linkers and adjuvants were used to fabricate the vaccine. Additionally, the designed mRNA vaccine was estimated to have an MW of 165023.50 Da and was found to be highly soluble. Assessing the solubility of a recombinant protein in overexpressed E. coli is of utmost importance for a diverse range of biochemical and functional experiments [80, 129]. The vaccine’s theoretical pI is 9.45, suggesting acidic characteristics. It also has instability and aliphatic indexes of 23.94 and 79.02, respectively. These values indicate that the vaccine exhibited hydrophobic characteristics consistent with the reported presence of aliphatic side chains. Vaccine development relies heavily on understanding protein folding into secondary and tertiary structures [80, 130]. Structural antigens, such as those in regions of unfolded protein and α-helical coils, have been crucial for eliciting protein-specific immune responses. Antibodies formed in response to opportunistic infections can bind to these two structural antigens if they refold into their native shape. Refining the vaccine led to a notable improvement in its tertiary structure, unveiling key features on the Ramachandran plot. The Ramachandran plot analysis revealed that a substantial majority (90.6%) of the vaccine residues are in preferred areas. Besides, a significant portion of the residues (7.05%) are located in the allowed regions, with a smaller fraction (0.8%) found in the generously allowed region. Based on the findings, the vaccine model’s overall quality is satisfactory. A docking study using human TLR-2 and TLR-4 evaluated the interaction between the vaccine and TLRs on immune cells. Subsequently, the docking analysis suggested that the vaccine had a significant level of affinity for TLR-2 and TLR-4 receptors. The MM-GBSA analysis of the "Vaccine—TLR-2" and "Vaccine—TLR-4" complexes also suggest a strong binding affinity with a free binding energy score of -141.07 (kcal/mol) and -271.72 (kcal/mol), respectively.
Codon optimization was performed to improve the expression of our recombinant vaccine in E. coli, especially the K12 strain, which provides a high degree of expression of the vaccine in bacteria with a GC content of 47.04% and CAI score of 1.0 (acceptable range for GC content is 30–70%, and for CAI is 1.0). The appearance of memory B-cells and T-cells was also observed, along with the persistent immunity of B-cells over one year. The activation of Th and subsequent production of IFN-γ and IL-2 exhibited distinctive features, evidenced by the immediate rise in IFN-γ and IL-2 concentrations after the first administration and their persistent elevation at maximum levels with repeated exposure to the antigen. This finding suggests an increase in the levels of Th cells and the production of IgM and IgG, which indicates a humoral immune response. Also, the minimum free energy of the mRNA vaccine was predicted to be -1760.00 kcal/mol, indicating the stability of the vaccine following its entry, transcription, and expression in the host [85, 87–90, 131].
5. Conclusion
In silico-based mRNA pancreatic cancer vaccines represent a promising and innovative approach to cancer immunotherapy. The vaccines are designed using computational methods to identify and encode tumor-specific antigens into mRNA molecules, which can be delivered to the patient’s immune cells to stimulate a robust anti-cancer immune response. The developed mRNA vaccine appeared to be soluble, hydrophilic, and acidic. The structural analysis revealed that the vaccine was a stable and functioning protein. Following that, the docking study indicated that the vaccine has a high affinity for TLR-2 and TLR-4 receptors, whereas the MM-GBSA analysis validated the statement. The vaccine was also firmly expressed in a computationally designed bacterial vector. Regarding immunological responses, the vaccine showed both humoral and adaptive immunity. Finally, this mRNA vaccine would be stable enough after its entrance, transcription, and expression in the host. The findings from these studies provide valuable insights into the properties and potential applications of a successful computationally designed PDAC vaccine. The development of this vaccine marks a significant milestone in the field of PDAC research and therapeutic advancements.
Supporting information
S1 FigThe secondary structure of the vaccine was predicted by the PSIPRED server, with the features (A) and types (B) of amino acids of the vaccine. In the sequence, among 1479 amino acids, where most of the amino acids were in the coil structure (grey), lesser in the helix structure (pink color), and least in the strand (yellow color) (A). The different types of amino acids in the sequence have been exhibited: the small nonpolar amino acids were predominant (orange), the hydrophobic amino acids (green) and the polar amino acids (red) were less prominent, and the aromatic plus cysteine residues (sky blue) were least prominent (B).(TIF)
S2 FigThe MM-GBSA free binding energy analysis of the “Vaccine—TLR-2" and "Vaccine—TLR-4" complexes.(TIF)
S3 FigA mountain plot representation of the MFE structure, the thermodynamic ensemble of RNA structures, and the centroid structure.(TIF)
S1 TableThe interactions between the vaccine and TLRs.(DOCX)
S2 TableThe discontinuous B-cell epitopes of the vaccine by Ellipro server.(DOCX)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adamska A, Domenichini A, Falasca M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int J Mol Sci. 2017;18(7):1338. doi: 10.3390/ijms 18071338 28640192 PMC 5535831 · doi ↗ · pubmed ↗
- 2Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–21. doi: 10.1158/0008-5472.CAN-14-0155 24840647 · doi ↗ · pubmed ↗
- 3Nguyen YTK, Park JS, Jang JY, Kim KR, Vo TTL, Kim KW, et al. Structural and Functional Analyses of Human Cha C 2 in Glutathione Metabolism. Biomolecules. 2019;10(0031). doi: 10.3390/biom 10010031 31878259 PMC 7022552 · doi ↗ · pubmed ↗
- 4Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. The Lancet. 2016;388(10039):73–85. doi: 10.1016/S 0140-6736(16)00141-0 26830752 · doi ↗ · pubmed ↗
- 5Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2(1):1–22. doi: 10.1038/nrdp.2016.22 27158978 · doi ↗ · pubmed ↗
- 6Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol. 2018;15(6):333–48. doi: 10.1038/s 41575-018-0005-x 29717230 · doi ↗ · pubmed ↗
- 7Golcher H, Brunner TB, Witzigmann H, Marti L, Bechstein W-O, Bruns C, et al. Neoadjuvant chemoradiation therapy with gemcitabine/cisplatin and surgery versus immediate surgery in resectable pancreatic cancer: results of the first prospective randomized phase II trial. Strahlenther Onkol. 2015;191(1):7–16. doi: 10.1007/s 00066-014-0737-7 25252602 PMC 4289008 · doi ↗ · pubmed ↗
- 8Huff AL, Jaffee EM, Zaidi N. Messenger RNA vaccines for cancer immunotherapy: progress promotes promise. J Clin Invest.132(6):e 156211. doi: 10.1172/JCI 156211 35289317 PMC 8920340 · doi ↗ · pubmed ↗