Chromosome-level genome assembly of an oligophagous leaf beetle Ophraella communa (Coleoptera: Chrysomelidae)
Yi-Ting Wang, Yan Zhang, Chao Ma, Wei-Hua Ma, Li-Jun Cao, Jin-Cui Chen, Wei Song, Jing-Fang Yang, Xu-Yuan Gao, Hong-Song Chen, Zhen-Ya Tian, Nicolas Desneux, Shu-Jun Wei, Zhong-Shi Zhou

TL;DR
Researchers created a high-quality, chromosome-level genome assembly for the leaf beetle Ophraella communa, which is used to control ragweed.
Contribution
The study provides the first chromosome-level genome assembly for Ophraella communa using multiple sequencing technologies.
Findings
The final genome assembly is 733.1 Mb, spanning 17 chromosomes with a contig N50 of 7.05 Mb.
The genome contains 25,873 protein-coding genes and numerous non-coding RNA sequences.
Repetitive elements make up 57.76% of the genome, totaling 414.41 Mb.
Abstract
The leaf beetle Ophraella communa LeSage (Coleoptera: Chrysomelidae) is an effective biological control agent of the common ragweed. Here, we assembled a chromosome-level genome of the O. communa by combining Illumina, Nanopore, and Hi-C sequencing technologies. The genome size of the final genome assembly is 733.1 Mb, encompassing 17 chromosomes, with an improved contig N50 of 7.05 Mb compared to the original version. Genome annotation reveals 25,873 protein-coding genes, with functional annotations available for 22,084 genes (85.35%). Non-coding sequence annotation identified 204 rRNAs, 626 tRNAs, and 1791 small RNAs. Repetitive elements occupy 414.41 Mb, constituting 57.76% of the genome. This high-quality genome is fundamental for advancing biological control strategies employing O. communa.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
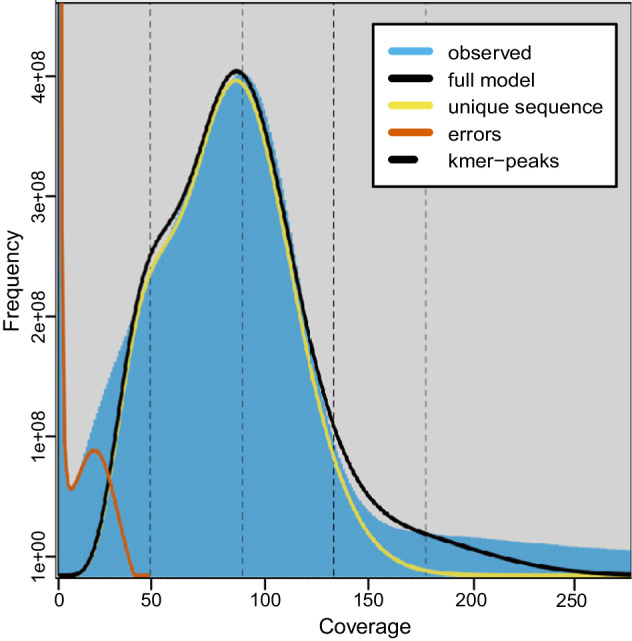 Figure 1
Figure 1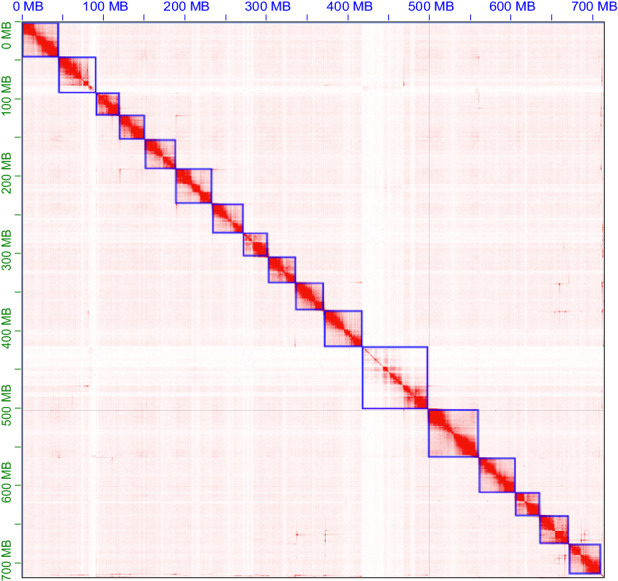 Figure 2
Figure 2- —The Program of Beijing Academy of Agriculture and Forestry Sciences (JKZX202208).
- —the National Key Research and Development Program of China (2023YFE0104800; 2022YFC2601400), the National Natural Science Foundation of China (32172494)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Biological Control of Invasive Species · Insect symbiosis and bacterial influences
Background & Summary
The leaf beetle Ophraella communa LeSage (Coleoptera: Chrysomelidae) is a native to North America^1^. It has been identified as a biological control agent of the common ragweed, Ambrosia artemisiifolia, a harmful invasive weed. It has achieved great success in controlling A. artemisiifolia spread and damage in different regions worldwide^2–4^. O. communa is an oligophagous insect feeding on various plants of the Asteraceae family and poses no threat to commercial crops. This beetle has short developmental periods, high fertility, and a long lifetime. The larvae and adults of O. communa can completely defoliate a common ragweed within a few insect generations^5^. To better apply this beetle, studies on its chemical ecology^6–8^, reproductive biology^9–11^, and cold tolerance genetics^12^ have been ongoing. Bouchemousse, et al.^13^ assembled a draft genome for O. communa on scaffold level^14^. A high-quality assembled and annotated genome is essential to assess potential adaptive processes by identifying underlying genetic mechanisms.
To this end, we applied Nanopore long-read, Illumina short-read sequencing, and High-throughput chromosome conformation capture technologies (Hi-C) to generate the first chromosome-level genome of O. communa. The assembled genome consists of a total scaffold length of 733.1 Mb, mapping to 17 chromosomes. Compared to the published contig version^14^, the contig N50 increased from 195.5 Kb to 7.05 Mb. A total of 414.41 Mb repeat sequences representing 57.76% of the whole genome were identified. Among these repeat sequences, 15.51% were classified as known repeat elements. We then performed structural and functional annotation on the obtained genome, incorporating transcriptome data from all developmental stages of O. communa. As the first chromosome-level genome assembly in the genus Ophraella, this high-quality reference genome not only provides information for better improvement of the biological control potential of O. communa, but also serves as a valuable resource for understanding the genetics, ecology, and evolution of Ophraella beetles.
Methods
Sample preparation and genomic DNA sequencing
A population of the O. communa collected from Guangxi, China, was established in the laboratory at the Institute of Plant Protection, Chinese Academy of Agricultural Science. This inbred population was fed with common ragweed for approximately ten generations in the laboratory under the following conditions: temperature of 27 ± 1 °C, relative humidity of 70 ± 5%, and a photoperiod of 14 L:10D. All the samples used in this study were from this inbred population which shared the almost same genetic background. Due to the small size of the O. communa pupa and its high fat content, the extracted DNA from one pupa is insufficient to conduct multiple sequencing methods. Thus, one pupa was used for the Nanopore library and another for Illumina library construction. The genomic DNA was extracted using the CTAB method. After removing the pupal shell, epidermis, and extracting as much fat body tissue as possible, the remaining tissue was homogenized in CTAB extraction buffer (20 g/L CTAB; 1.4 mol/L NaCl; 0.1 mol/L Tris-HCl; 20 mmol/L Na_2_EDTA). Then, the genomic DNA was purified using a Blood and Cell Culture DNA Midi Kit (QIAGEN, Germany). The purity of the extracted DNA was assessed through 0.75% agarose gel electrophoresis, while the concentration was assessed using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, USA). Illumina paired-end (PE) library featuring an insert size of approximately 350 bp utilized the TruSeq Nano DNA HT Sample Preparation Kit (Illumina, San Diego, California, USA). Subsequently, paired-end reads of 150 bp were generated on the Illumina NovaSeq 6000 platform. The output amounted to 83.00 GB of clean data, providing coverage of 115.67 × . The G + C content was measured to be 33.32%, and the peak insert size observed was 169 bp. After filtering using fastp version 0.23.4^15^, 82.13 GB of data remained, with 76.63 GB (93.08%) of high-quality sequences exceeding Q30 threshold (Table 1). The long-insert library was constructed with the SQK-LSK108 1D Ligation Sequencing Kit (Oxford Nanopore Technologies, Kidlington, Oxford, UK) using the genomic DNA. The long-insert library underwent sequencing on the Nanopore PromethION sequencer at GrandOmics (Wuhan, China). A total of 105.86 Gb (147.54 × coverage) of long-reads were generated, with a mean Q score of 11.50. The N50 length of clean data is 274,07 bp, the longest reads is 465963 bp, and the average length is 16,445 bp (Table 1).Table 1. Library sequencing data and methods used in this study to assemble the Ophraella communa genome.Sequencing strategyPlatformUsageInsertion sizeClean data (Gb)Coverage (X)Short-readsIlluminaGenome survey350 bp83.00115.67Long-readsNanoporeAssembly12–20 kb105.86147.54Hi-CIlluminaHi-C assembly350 bp42.5659.33RNA-seqIlluminaAnnotation350 bp115.13157.04
Hi-C library preparation and sequencing
One pupa with unknown sex was used to create the Hi-C library to capture genome-wide chromatin interactions. After washing the surface with PBS buffer, the pupal shell, epidermis and as much fat body tissue as possible were removed. Subsequently, cross-linking was performed using a 2% formaldehyde isolation buffer, followed by treatment with the restriction enzyme MboI to digest chromatin. The Hi-C samples were then extracted via biotin labeling and flat-end ligation, after which the ligated DNA was fragmented into 350 bp fragments. Subsequently, the Hi-C library was sequenced on the Illumina NovaSeq platform with paired-end 150-bp reads. The Hi-C Illumina sequencing generated 42.56 Gb (59.33 × coverage) of clean data (Table 1). The G + C content was measured to be 33.50%, and the mean length of reads was 100 bp. After filtering using fastp version 0.23.4^15^, 42.48 GB of data remained, with 40.95 GB (96.39%) of high-quality sequences exceeding Q30 threshold.
Transcriptome sequencing
To obtain comprehensive genome annotation, we conducted transcriptome sequencing on different stages of O. communa, including two adult pooled samples, two egg pooled samples, two pupa pooled samples, and three larva pooled samples. Each adult pooled sample consisted of five male and female adults of O. communa. Each egg sample consisted of approximately 100 eggs. Each pupa and larvae samples consisted of five pupae and larvae, respectively. The total RNA was extracted with the TRIzol reagent (Thermo Fisher Scientific, USA). Except for the pupae after removing the pupal shell, all other samples are homogenized using the entire organism with TRIzol. cDNA was synthesized from total RNA using PrimeScript™ RT reagent kit with gDNA Eraser (Perfect Real Time; Takara, Japan) following the manufacturer’s instructions. Then the cDNA was constructed to the paired-end libraries using the VAHTSTM mRNA-seq v2 Library Prep Kit (Vazyme, Nanjing, China) with an insert size of approximately 350 bp and then sequenced on the Illumina NovaSeq 6000 platform with paired-end reads of 150 bp. The nine libraries generated 115.13 GB of clean data, providing coverage of 157.04 × . The mean G + C content was measured to be 38.82%, and the mean peak insert size observed was 265 bp. After filtering using fastp version 0.23.4^15^, 114.47 GB of data remained, with 96.89 GB (93.64%) of high-quality sequences exceeding Q30 threshold (Table 1).
Estimation of genomic characteristics
The genome features of O. communa were surveyed using the k-mer method based on Illumina short reads. The k-mer count histogram was generated using Jellyfish version 2.2.10 with the following parameters: ‘count -m 25 -C -s 5 G’^16^. We used GenomeScope^17^ version 1.0 to estimate the genome size, heterozygosity, and duplication rate. The analysis based on 25-mers estimated the genome size of O. communa to be approximately 741.69 Mb, showing a high degree of duplication (4.9%) and heterozygosity (0.73%) (Fig. 1).Fig. 1. The estimated characteristics of Ophraella communa genome based on Illumina short-read data using 25-mers count histogram. Genome size was estimated to be 741.69 Mb, with a duplication rate of 4.9% and heterozygosity rate of 0.73%.
Genome assembly
The Nanopore long reads were corrected and assembled into contigs using NextDenovo version 2.5.0 (https://github.com/Nextomics/NextDenovo) with parameters: ‘read_cutoff = 1k, genome_size = 750 m, pa_correction = 20, nextgraph_options = -a 1’. Subsequently, the contigs assembly underwent three rounds of polishing using NextPolish version 1.4.0 18^18^, incorporating the parameters: ‘genome_size = auto, sgs_options = -max_depth 100 -bwa, rerun = 3’. To generate a high-quality chromosome-scale genome, the Hi-C data were initially mapped to the contig assembly using BWA-MEM version 0.7.17^19^ with the parameters:‘mem -SP5M’. Then, the MboI sites were generated using the’generate_site_positions.py’script in the Juicer^20^ version 1.6 with default parameters. 3D-DNA (3D de novo assembly, version 180114) pipeline^21,22^ was employed to order, orient, and cluster the contigs into scaffolds with a modified parameter of ‘–editor-repeat-coverage 5, -r 2’. To achieve the final chromosome assembly, manual scaffold ordering was performed using Juicebox version 1.11.08 (https://github.com/aidenlab/Juicebox).
At the contig level, the genome is 735.31 Mb, comprising 220 contigs, with a contig N50 of 7.05 Mb (Table 2). At the chromosomal level, the resulting genome of O. communa measures 733.13 Mb, organized into 17 scaffold groups, with a scaffold N50 of 45.03 Mb (Table 2). The karyotype of O. communa is 2n = 18, consisting of one pair of heteromorphic sex chromosomes (XX in females, XY in males) and 17 pairs of autosomes^13^, which were well-distinguished from the chromatin interaction heatmap (Fig. 2). The scaffold groups vary in length, with the longest group spanning 81.99 Mb and the shortest group spanning 29.41 Mb. The O. communa genome has a G + C content of approximately 31.98% (Table 2).Table 2. Features of Ophraella communa genome.Genome featureValue (bp)Total length (bp)733,128,312Contigs N50 (bp)7,051,505Scaffold N50 (bp)45,027,602Longest scaffold length (bp)81,994,011G + C (%)31.98Anchored to chromosome (Mb, %)717,507,083 (97.87%)Fig. 2. Genome-wide all-by-all Hi-C interaction identified 17 pseudo-chromosome linkage groups of Ophraella communa.
Repeat element and non-coding RNA annotation
The assembled genome was analyzed for repetitive elements and transposable element families using RepeatMasker version 4.0.7^23^ against the Insecta repeats within RepBase Update (http://www.girinst.org) with parameters of ‘-e ABBlast, -species Insecta’. Additionally, ab initio prediction was conducted using the program RepeatModeler version open-1.0.8 (https://www.repeatmasker.org/RepeatModeler/). Non-coding RNAs (ncRNAs) were annotated by aligning the genomic sequence against RFAM (http://rfam.xfam.org/) using version 1.1.2^24^ with the parameter of ‘-e ABBlast’. The prediction of transfer RNA (tRNA) was conducted through tRNAscanSE v.1.3.1^25^ with default parameters. For ribosomal RNA (rRNA) prediction, RNAmmer-1.2^26^ was employed with parameters: ‘-S euk, -multi’.
The repetitive elements constituted 414.41 Mb (57.76%) of the O. communa genome, with 15.51% being classified as known repeat elements (Table 3). According to the Rfam databases, our predictions for the O. communa genome revealed 204 rRNAs, 626 tRNAs, and 1791 small RNAs (Table 4).Table 3. Repeats elements statistics in genome of Ophraella communa.ItemsNumber of elementsLength occupied (bp)Percentage of sequence (%)Retroelements139619570135047.95SINEs198122130.00LINEs107673318683924.44LTR elements31748251328993.50DNA transposons129099399169805.56Total interspersed repeatsNA9762841013.61Small RNA17914183460.06Satellites1320340.00Simple repeats192320113239581.58Low complexity5209426926210.38Note: SINEs, short interspersed nuclear elements; LINEs, long interspersed nuclear elements; LTR, long terminal repeat.Table 4. Statistics of non-coding RNAs in Ophraella communa genome.ClassTypeNumberrRNA counts8s_rRNA9418S_rRNA5528s_rRNA55Candidate tRNAs read435Cove statsCove-confirmed tRNAs626Bases scanned by Cove556066tRNA CounttRNAs decoding Standard 20 AA348Selenocysteine tRNAs (TCA)0Possible suppressor tRNAs (CTA, TTA)0tRNAs with undetermined/unknown isotypes4Predicted pseudogenes274Total tRNAs626tRNAs with intron27
Gene and functional predictions
Protein-coding genes in the were annotated utilizing homolog-based, RNA-seq-based, and ab initio methods using Maker genome annotation pipeline version 3.01.04^27^. The transcriptome of O. communa was initially assembled by employing StringTie version 1.3.3b^28^ and PASA version 2.0.2^29^. This process utilized the FASTA files of the final chromosome assembly and transcriptome sequencing reads as input data, with default parameters. Ab initio prediction models were trained using homologous genes from Tribolium castaneum^30^ and the transcripts for Augustus version 3.4.0^31^ with default parameters and SNAP version 2006-07-28^32^ with the parameters of ‘-categorize 1000, -export 1000, -plus’. The results were utilized for subsequent rounds of model training and annotation. Three rounds of Maker annotations were conducted and improved by PASA. Then, this result was integrated with the result of a deep-learning structural gene annotations approach Helixer^33^ and then filtered based on gene expression evidence and functional annotation. In order to ensure the accuracy of the annotation results, genes with fragments per kilobase per million (FPKM) values equal to 0 were excluded for further analysis. The protein-coding genes, Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) items underwent annotation using eggNOG-Mapper version 2.1.9 within the Expected eggNOG DB version 5.0.2^34^. This process utilized specific parameters, including ‘–tax_scope auto’, ‘–go_evidence experimental’, ‘–target_orthologs all’, ‘–seed_ortholog_evalue 0.001’, ‘–seed_ortholog_score 60’, and ‘–override’. In the chromosome-level assembly, we annotated 25,873 protein-coding genes, which is closer to the number of genes found in related Coleoptera species and general insect genomes compared to the 75,642 protein-coding genes in the genome reported by Bouchemousse et al.^13^. In total, 25,873 protein-coding genes were annotated, with 22,084 genes (85.35%) being functionally annotated^35^.
Data Records
The O. communa genome project was deposited at NCBI under the BioProject accession number PRJNA899605. Genomic Illumina sequencing data are available in the Sequence Read Archive at NCBI under accession SRR27238374^36^. Hi-C sequencing data are available in the Sequence Read Archive at NCBI under accession number SRR27307846^37^. Genomic Nanopore sequencing data are available in the Sequence Read Archive at NCBI under accession number SRR27290278^38^. RNA-seq data are available in the Sequence Read Archive at NCBI under accession number SRR27334077-SRR27334085^39–47^. The final chromosome assembly was deposited in GenBank at NCBI under accession number GCA_035357415.1^48^. The genome annotation files are available in Figshare under a DOI of 10.6084/m9.figshare.24901596.v1^35^.
Technical Validation
The accuracy of the final genome assembly was assessed by aligning Illumina short reads and RNA-seq data to the O. communa genome using BWA-MEM2 version 2.2.1 (https://github.com/lh3/bwa). The analysis revealed that mapping rate of 99.56% for the short reads to the genome. The mapping rates for the respective stages-specific transcriptomic data ranged from 89.39% to 94.12%.
In evaluating the completeness of the O. communa genome, an analysis was conducted using BUSCO version 5.2.2^49^ with the insecta-odb10 database, which consists of 1,367 genes. The BUSCO analysis revealed that 99.7% of the evaluated single-copy genes at the contig level were determined to be complete (96.6% single-copy genes and 3.1% duplicated genes). At the chromosome level, it was observed that 99.7% of the assessed single-copy genes were classified as complete (97.1% single-copy genes and 2.6% duplicated genes). For all protein-coding genes and functionally annotated protein-coding genes, it was determined that 95.1% of them were identified as complete (92.7% single-copy genes and 2.4% duplicated genes) (Table 5).Table 5. Completeness of the assembled genomes and sets of protein-coding genes evaluated by BUSCO analysis.DataComplete gene%Single-copied gene%Duplicated gene %Fragmented gene%Missing gene%Contig level assembly99.796.63.10.10.2Chromosome level assembly99.797.12.60.10.2All protein-coding gene95.192.72.42.52.4Functionally annotated protein-coding gene95.192.72.42.52.4
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Zhou ZS Guo JY Ai HM Li M Wan FH Rapid cold-hardening response in Ophraella communa Le Sage (Coleoptera: Chrysomelidae), a biological control agent of Ambrosia artemisiifolia LBIOCONTROL SCI TECHN 20112121522410.1080/09583157.2010.534549 · doi ↗
- 2Schaffner U Biological weed control to relieve millions from Ambrosia allergies in Europe Nat. Commun 202011174510.1038/s 41467-020-15586-132317698 PMC 7174423 · doi ↗ · pubmed ↗
- 3Guo J-Y Control efficiency of leaf beetle, Ophraella communa, on the invasive common ragweed, Ambrosia artemisiifolia, at different growing stages BIOCONTROL SCI TECHN 2011211049106310.1080/09583157.2011.603823 · doi ↗
- 4Essl F Biological Flora of the British Isles: Ambrosia artemisiifolia J. Ecol.20151031069109810.1111/1365-2745.12424 · doi ↗
- 5Yamanaka T Tanaka K Otuka A Bjørnstad ON Detecting spatial interactions in the ragweed (Ambrosia artemissifolia L.) and the ragweed beetle (Ophraella communa Le Sage) populations ECOL RES 20062218519610.1007/s 11284-006-0300-9 · doi ↗
- 6Ma C Olfactory co-receptor is involved in host recognition and oviposition in Ophraella communa (Coleoptera: Chrysomelidae)INSECT MOL BIOL 20202938139010.1111/imb.1264332291884 · doi ↗ · pubmed ↗
- 7Ma C Ocom CSP 12, a chemosensory protein expressed specifically by ovary, mediates reproduction in Ophraella communa (Coleoptera: Chrysomelidae)Front Physiol 201910129010.3389/fphys.2019.0129031681004 PMC 6803423 · doi ↗ · pubmed ↗
- 8Ma C Identification of candidate chemosensory genes of Ophraella communa Le Sage (Coleoptera: Chrysomelidae) based on antennal transcriptome analysis Sci Rep 201991555110.1038/s 41598-019-52149-x 31664149 PMC 6820725 · doi ↗ · pubmed ↗