Validation and depth evaluation of recurrent neural network‐based ultra low‐pass genome sequencing for the detection of absence of heterozygosity: A multi‐centre study of 409 cases
Yeqing Qian, Jianjun Zhu, Zhiguo Tang, Yan Sun, Zhonghua Wang, Fei Tang, Yun Yang, Linlin Fan, Yixi Sun, Bei Liu, Min Chen, Yuqin Luo, Junjie Hu, Kai Yan, Jianfen Man, Lina Wang, Cangcang Jia, Ping Tang, Xinyi Zhu, Chaohong Wang, Junxiang Tang, Yuanyuan Xia, Xueqin Guo

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
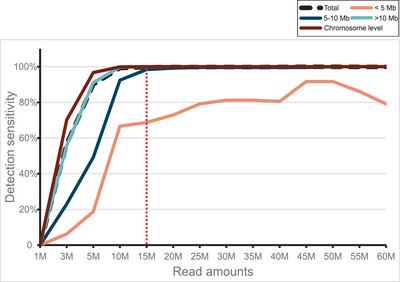 Figure 1
Figure 1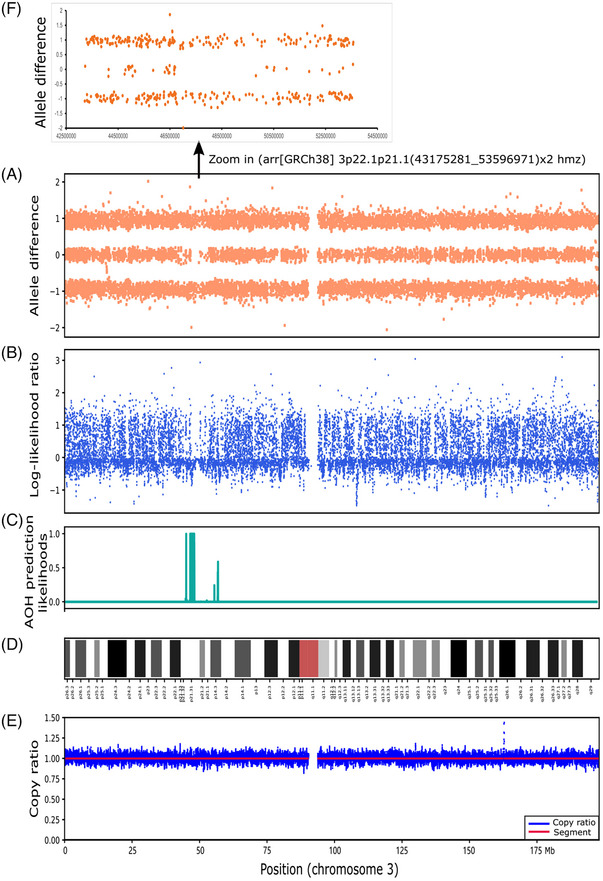 Figure 2
Figure 2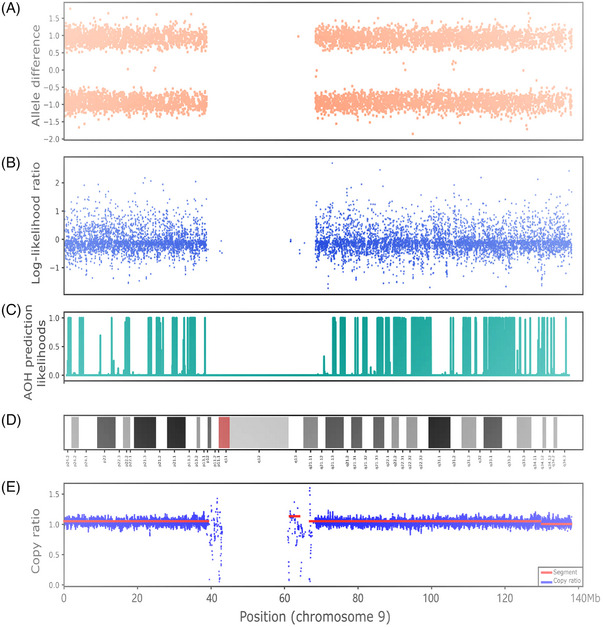 Figure 3
Figure 3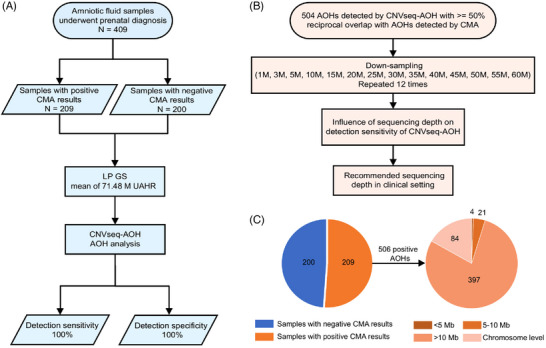 Figure 4
Figure 4- —National Key R&D Program of China 10.13039/501100012166
- —4+X Clinical Research Project of Women’s Hospital, Zhejiang University School of Medicine
- —Technology Bureau of Jiaxing, Zhejiang Province
- —Anhui Key Research and Development Program
- —Zhejiang Provincial Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomic variations and chromosomal abnormalities · Prenatal Screening and Diagnostics · Genetic Syndromes and Imprinting
Dear Editor,
We conducted a comprehensive clinical assessment of our newly developed method, CNVseq‐AOH, for the detection of absence of heterozygosity (AOH) using low‐pass genome sequencing (LP GS) with ultra‐low sequencing data in this multi‐centre study.
Although AOH in chromosomes does not necessarily have clinical consequences, the detection of AOH is clinically important when it is related to imprinting effects or autosomal recessive disease mechanisms.1, 2, 3, 4 LP GS (also known as CNVseq) has enabled the detection of copy‐number variants (CNVs) for its superiority in sensitivity and specificity.2, 5, 6 In China, it has been recommended as a first‐line diagnostic method for foetuses displaying structural abnormalities.7 However, the investigation on the use of LP GS for the detection of AOH is limited, and furthermore, LP GS has never been reported for the detection of AOH in a low‐pass setting of less than 1‐fold. To establish a comprehensive clinical assessment of our newly developed method (https://github.com/helplessness/CNVseq‐AOH), CNVseq‐AOH, for the detection of AOH, and to investigate the optimal sequencing depth, we gathered 409 samples (by far the largest clinical samples) of amniotic fluid from three hospitals. Our data showed that CNVseq‐AOH in large‐scale clinical practice maintained high sensitivity (100%) and specificity (100%), providing good evidence for its clinical application. This multi‐centre study demonstrated the feasibility of CNVseq‐AOH for the detection of AOH in real clinical settings.
First, using samples with positive AOH regions from the 1000 Genomes Project (1KGP), we performed concordant analysis for CNVseq‐AOH and chromosomal microarray analysis (CMA). CNVseq‐AOH was aimed to predict AOH using LP GS with ultra‐low sequencing data. Compared to CMA, CNVseq‐AOH showed high sensitivity and great potential to improve the genetic testing of AOH.
Second, to assess and comprehensively study the performance of CNVseq‐AOH in a real clinical environment, a total of 409 archived DNA samples of amniotic fluid (209 with positive AOH results (Table S1) and 200 samples with negative AOH results by CMA (Table S3)) from Women's Hospital, Zhejiang University School of Medicine, Jiaxing Maternity and Child Health Care Hospital and Anhui Province Maternity & Child Health Hospital from April 2017 to March 2023, were recruited. Among the 409 samples, CMA identified 209 cases with positive AOHs (506 AOH regions) (Figure 1). LP GS for the 409 samples were conducted on the MGISEQ‐2000 platform for single‐end (35 bp) sequencing as previously described.8 For these samples, an average of 71.48 M uniquely aligned high quality reads (UAHRs) were obtained, approximately 0.83‐fold for each sample. After sequencing, AOH detection was performed for each sample using CNVseq‐AOH.
The results of CMA were blinded to individuals who were analysing using CNVseq‐AOH. When using the CMA results as a reference, the diagnostic yield of CNVseq‐AOH was found to be equivalent to that of CMA (Table S1). Specifically, CNVseq‐AOH demonstrated a sensitivity of 100% (209/209) and a specificity of 100% (200/200) in our cohort.
The overlap for the 506 AOH regions detected by CMA and CNVseq‐AOH was further analysed (Table S1). It showed that ∼99.60% (504/506) of the AOHs detected by CNVseq‐AOH had a reciprocal overlap of more than 50% with the AOHs detected by CMA (Table S2). Compared with CMA, two AOHs were detected to be with an overlap of less than 50% in case PS201 (Figure 2) and case PS44 (Figure 3). Case PS201 included 1 positive AOH across the whole chromosome 9 by CMA. In the CNVseq‐AOH detection results, the region was divided into multiple subregions (Figure 2C), with a total overlap of less than 50% between the two methods. Further analysis revealed low‐level mosaicism (∼6.7%) of the whole chromosome 9 (Figure 2E). The presence of this mosaicism can affect the performance of CNVseq‐AOH, leading to the discrepancy observed between the two methods. For the ∼10.4 Mb AOH in case PS44, abnormal signals were detected in the reported regions of CMA (Figure 3C), with some regions showing relatively dispersed signals from allele difference analysis (Figure 3F). These differences may be due to the differences in detection principles between the two methods.
Third, to examine how the detection sensitivity of CNVseq‐AOH is affected by sequencing depth, depth evaluation was performed (Figure 1B). In total, the UAHRs for 504 AOHs were utilized to create downsampling samples (14 different sequencing depths for each sample). As a result, the performance of CNVseq‐AOH in downsampling samples varied depending on the size of the AOH (Figure 4 and Table S2). The detection sensitivity of 4 AOHs (cases PS1‐4) with sizes less than 5 Mb was greatly influenced by UAHR. The detection sensitivity of AOHs with sizes between 5 and 10 Mb was also influenced by UAHR (Figure 4) and reached a plateau at 20 M UAHR. For AOHs larger than 10 Mb and at the chromosome level, the impact of sequencing depth becomes less pronounced (Figure 4 and Table S2), reaching 99.74% and 99.89% at 10 M UAHR. Overall, the detection sensitivity tended to increase as the number of UAHRs increased, and it reached a plateau at 15 M UAHRs for all the 504 AOHs (Figure 4). When using 15 M UAHRs, the overall detection sensitivity is over 99.66%. Therefore, 15 M UAHRs were considered optimal for detecting AOHs using CNVseq‐AOH based on our cohort, approximately 23 times (342.86 M reads) less than the existing method.9
What is more, CNVseq‐AOH addressed the long‐standing challenge of standard LP GS in detecting AOH. Typically, the cost of CMA for one sample is more than 600.[10](#ctm21752-bib-0010) In our laboratory, the overall cost of LP GS for a single case was approximately 248.8 Combining CNVseq‐AOH, LP GS could achieve the same level of performance as CMA for AOH detection (> 10 Mb) at a very low sequencing depth.
In summary, we developed a method for predicting AOH using LP GS data and tested its performance in this multi‐centre study. CNVseq‐AOH can identify regions of AOH accurately in the clinical setting and possesses great potential to improve the genetic testing of AOH. According to these findings, CNVseq‐AOH has demonstrated high precision in detecting AOHs, indicating its promising application in clinical settings.
AUTHOR CONTRIBUTIONS
Suping Li, Lijie Song, Jiansheng Zhu, Minyue Dong, Yeqing Qian, Jianjun Zhu, Zhiguo Tang and Yan Sun contributed to the conception and design of the study. Yan Sun wrote the first draft of the article. Yun Yang, Linlin Fan, Yixi Sun, Bei Liu, Min Chen, Yuqin Luo, Junjie Hu and Kai Yan designed and performed the experiments. Yan Sun, Zhonghua Wang, Fei Tang, Jianfen Man, Lina Wang, Cangcang Jia, Ping Tang, Xinyi Zhu, Chaohong Wang, Junxiang Tang, Yuanyuan Xia, Xueqin Guo, Kang Zhang and Xiaoli Wang performed data analysis. Yeqing Qian, Lijie Song, Minyue Dong and Yan Sun contributed to revising the manuscript. All authors reviewed the manuscript and approved the submitted version.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
FUNDING INFORMATION
This study was supported by the National Key R&D Program of China (2023YFC2705600). This work was also supported by the 4+X Clinical Research Project of Women's Hospital, Zhejiang University School of Medicine (ZDFY2023‐4XPY201), Technology Bureau of Jiaxing, Zhejiang Province (2023AY31030), Anhui Key Research and Development Program (2022e07020031), and Zhejiang Provincial Natural Science Foundation of China (LY22H110004). These projects are non‐profit research projects by the government and had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
ETHICS STATEMENT
This study was approved by the Institutional Review Board of Women's Hospital, Zhejiang University School of Medicine (NO. IRB‐20230313‐R), Jiaxing Maternity and Child Health Care Hospital (NO. 2023−047), Anhui Province Maternity & Child Health Hospital (NO. 2023‐005‐01) and the Institutional Review Board of BGI (NO. BGI‐IRB 23140).
Supporting information
Supporting Information
Supporting Information
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Yauy K , de Leeuw N , HG Yntema , Pfundt R , Gilissen C . Accurate detection of clinically relevant uniparental disomy from exome sequencing data. Genet Med. 2020;22(4):803‐808.31767986 10.1038/s 41436-019-0704-x PMC 7118024 · doi ↗ · pubmed ↗
- 2Dong Z , Zhang J , Hu P , et al. Low‐pass whole‐genome sequencing in clinical cytogenetics: a validated approach. Genet Med. 2016;18(9):940‐948.26820068 10.1038/gim.2015.199 · doi ↗ · pubmed ↗
- 3Sahoo T , Dzidic N , Strecker MN , et al. Comprehensive genetic analysis of pregnancy loss by chromosomal microarrays: outcomes, benefits, and challenges. Genet Med. 2017;19(1):83‐89.27337029 10.1038/gim.2016.69 · doi ↗ · pubmed ↗
- 4Xiang J , Li R , He J , et al. Clinical impacts of genome‐wide noninvasive prenatal testing for rare autosomal trisomy. Am J Obstet Gynecol MFM. 2023;5(1):100790.36377092 10.1016/j.ajogmf.2022.100790 · doi ↗ · pubmed ↗
- 5Wang H , Dong Z , Zhang R , et al. Low‐pass genome sequencing versus chromosomal microarray analysis: implementation in prenatal diagnosis. Genet Med. 2020;22(3):500‐510.31447483 10.1038/s 41436-019-0634-7PMC 7042067 · doi ↗ · pubmed ↗
- 6Chau MHK , Wang H , Lai Y , et al. Low‐pass genome sequencing: a validated method in clinical cytogenetics. Hum Genet. 2020;139(11):1403‐1415.32451733 10.1007/s 00439-020-02185-9 · doi ↗ · pubmed ↗
- 7Clinical Genetics Group Of Medical Genetics Branch Chinese Medical A, Professional Committee For Prenatal Diagnosis Of Genetic Diseases Medical Genetics Branch Of Chinese Medical A, Group Of Genetic Disease P, Control Birth Defect P, Control Committee Of Chinese Society Of Preventive M . Expert consensus on the application of low‐depth whole genome sequencing in prenatal diagnosis. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2019;36(4):293‐296.30950010 10.3760/cma.j.issn.1003-9406.2019.04.001 · doi ↗ · pubmed ↗
- 8Qian Y , Sun Y , Guo X , et al. Validation and depth evaluation of low‐pass genome sequencing in prenatal diagnosis using 387 amniotic fluid samples. J Med Genet. 2023;60(10):933‐938.37012053 10.1136/jmg-2022-109112 · doi ↗ · pubmed ↗