Hypervalent iodine-catalyzed amide and alkene coupling enabled by lithium salt activation
Akanksha Chhikara, Fan Wu, Navdeep Kaur, Prabagar Baskaran, Alex M Nguyen, Zhichang Yin, Anthony H Pham, Wei Li

TL;DR
A new method uses lithium salts to activate hypervalent iodine catalysts, enabling precise coupling of amides and alkenes.
Contribution
A novel lithium salt activation strategy for hypervalent iodine catalysis is introduced.
Findings
Lithium salts enable activation of hypervalent iodine catalysts for intermolecular coupling.
Amides can be selectively added to activated olefins with high regioselectivity.
Abstract
Hypervalent iodine catalysis has been widely utilized in olefin functionalization reactions. Intermolecularly, the regioselective addition of two distinct nucleophiles across the olefin is a challenging process in hypervalent iodine catalysis. We introduce here a unique strategy using simple lithium salts for hypervalent iodine catalyst activation. The activated hypervalent iodine catalyst allows the intermolecular coupling of soft nucleophiles such as amides onto electronically activated olefins with high regioselectivity.
Click any figure to enlarge with its caption.
 Scheme 1
Scheme 1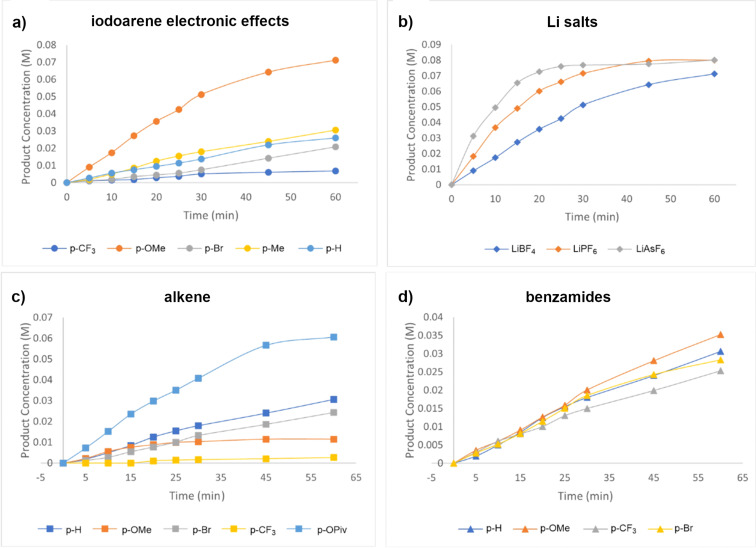 Figure 1
Figure 1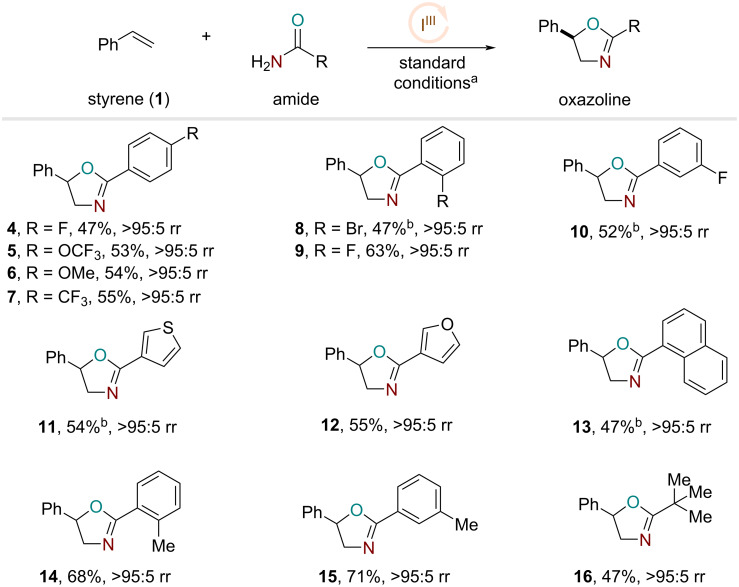 Figure 2
Figure 2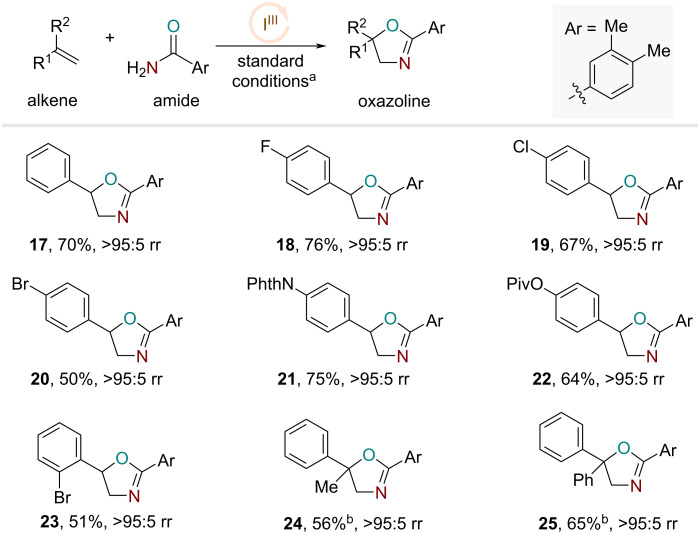 Figure 3
Figure 3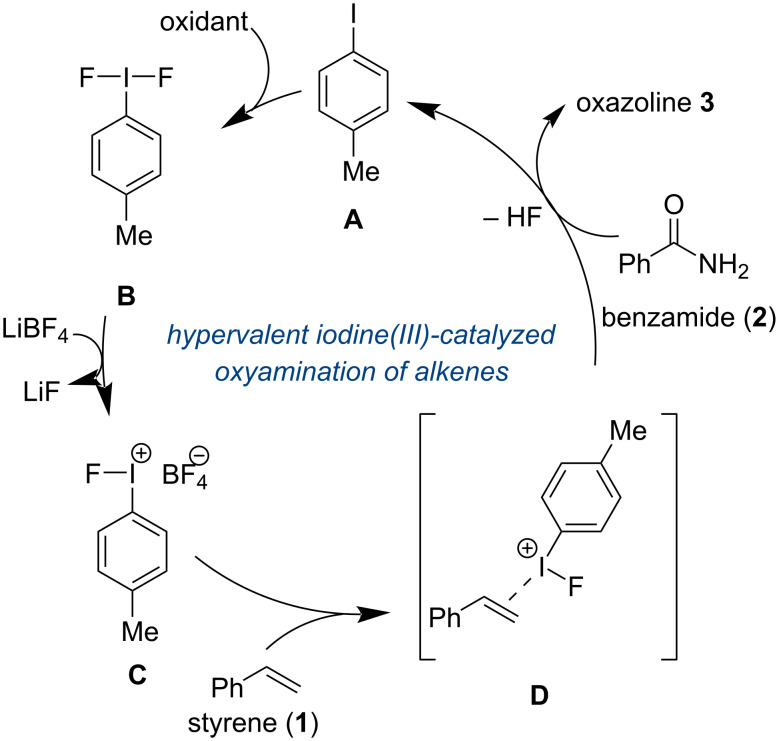 Figure 4
Figure 4|
| |||||
|
| |||||
| entrya | precatalyst (mol %) | solvent (M) | additive (mol %) | yield (%)c | rr |
|
| |||||
| 1 | MeNO2 (0.5) | LiBF4 (100) | 59 | >95:5 | |
| 2 | MeCN (0.5) | LiBF4 (100) | 41 | 94:6 | |
| 3 | MeOH (0.5) | LiBF4 (100) | 0 | – | |
| 4 | DMF (0.5) | LiBF4 (100) | 0 | – | |
| 5 | MeNO2 (0.3) | LiBF4 (100) | 62 | >95:5 | |
| 6b | MeNO2 (0.25) | LiBF4 (100) | 64 | >95:5 | |
| 7 | MeNO2 (0.25) | LiBF4 (100) | 65 (61) | >95:5 | |
| 8 | MeNO2 (0.5) | LiBF4 (100) | 55 | >95:5 | |
| 9 | MeNO2 (0.5) | LiPF6 (100) | 56 | >95:5 | |
| 10 | MeNO2 (0.5) | AgBF4 (100) | 12 | >95:5 | |
| 11 | – | MeNO2 (0.5) | LiBF4 (100) | 0 | – |
| 12d | MeNO2 (0.5) | LiBF4 (100) | 0 | – | |
| 13 | MeNO2 (0.5) | – | 8 | >95:5 | |
- —National Institute of General Medical Sciences of the National Institutes of Health
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsOxidative Organic Chemistry Reactions · Synthetic Organic Chemistry Methods · Advanced Synthetic Organic Chemistry
Introduction
Hypervalent iodine(III) reagents, also known as λ^3^–iodanes, have been well established and used in organic synthesis for the past decades [1–5]. The pioneering works of Fuchigami and Fugita, Ochiai, Kita, and later the development of chiral hypervalent iodines by Wirth, Kita, Ishihara, Muñiz, and many others, have firmly established these reagents as useful catalysts for a wide variety of chemical transformations [6–17]. A number of features, including low toxicity, high stability, ease of handling, and versatile reactivity, etc. render these catalysts highly attractive for adoption in organic synthesis. In particular, the field of olefin difunctionalization, known for its rapid assembly of molecular complexity, has been a fertile ground for innovation for hypervalent iodine catalysis, which often involves the catalytic use of an iodoarene with stoichiometric oxidants such as MCPBA, Selectfluor, etc. [18–20]. Earlier and recent hypervalent iodine-catalyzed olefin halofunctionalizations by several groups have predicated on the use of intramolecular olefin substrates tethered with a nucleophile to avoid the lack of regiochemical additions (Scheme 1a) [21–28]. Intermolecular hypervalent iodine-catalyzed olefin difunctionalizations have been realized for olefin dihalogenation, dioxygenation and diamination reactions, where often the same type of nucleophiles were incorporated (Scheme 1b) [29–40]. Intermolecular hypervalent iodine catalysis with the regioselective additions of two distinct nucleophilic functionalities across an olefin, however, remains challenging with limited solutions [41–46]. Notably, an interesting work by Hashimoto has recently enabled the intermolecular addition of N-(fluorosulfonyl)-protected carbamates as oxyamination reagents across a variety of olefin structures [47]. This work engages the hypervalent iodine catalyst in an anionic ligand exchange with the substrate, which then partitions into an ion pair suitable for olefin activation, followed by the addition of the bifunctional anionic carbamate (Scheme 1c).
Hypervalent iodine-catalyzed olefin difunctionalizations background.
Our hypothesis here aims to directly access the reactivity of the cationic hypervalent iodine catalyst through an initial activation first, which we reason will then enable soft nucleophiles such as unadorned amides to readily participate in the ensuing olefin addition. In this regard, we wondered if the hypervalent iodine with difluoro ligands could undergo salt metathesis with lithium salts such as LiBF_4_ or LiPF_6_ to afford the more reactive cationic hypervalent iodine catalyst. The cationic hypervalent iodine catalyst could then activate the olefin to allow the addition of bifunctional nucleophiles such as an amide to achieve an overall olefin oxyamination process. We have previously reported a series of iodide-catalyzed processes, in which the electrophilicity of the halogen source could be modulated to render different classes of nucleophiles for additions onto olefins in various olefin difunctionalization reactions [48–52]. In particular, we demonstrated that addition of either a Lewis acid or a base could activate amides to couple with alkenes regioselectively to furnish their respective oxazoline regioisomer (Scheme 1d). Herein, we report that lithium salts such as LiBF_4_ or LiPF_6_, which are often used in lithium-ion batteries, can be used to activate hypervalent iodine catalysts to enable olefin oxyamination reactions with simple bifunctional amide nucleophiles (Scheme 1e).
Results and Discussion
Our studies here focused on the development of hypervalent iodine-catalyzed amide and alkene coupling reaction [53–55]. In this case, we started with styrene (1) and benzamide (2) as the standard substrates. Using iodotoluene A as the hypervalent iodine catalyst precursor, Selectfluor as the oxidant, and LiBF_4_ as the lithium salt for hypervalent iodine activation, we were gratified to observe the formation of the desired oxazoline 3 in 59% yield as the major regioisomer in nitromethane (MeNO_2_) solvent (Table 1, entry 1). To further improve the reaction efficiency, we screened several additional parameters including solvents and concentration. In these cases, we found that while both acetonitrile and MeNO_2_ (0.25 M) were suitable solvents, other solvents in general afforded no product formation (Table 1, entries 2–6). Lower catalyst loading and longer reaction time did not improve the overall reaction efficiency (Table 1, entries 6–8). Furthermore, we evaluated several salt additives containing different counterions, and found that LiBF_4_ was the optimal additive (Table 1, entries 7, 9, and 10). The optimal conditions were shown in entry 7 in Table 1, resulting in the formation of the desired oxazoline product 3 in 61% isolated yield. Control reactions in the absence of either the precatalyst or oxidant afforded no product formation (Table 1, entries 11 and 12). The control reaction in the absence of the lithium salt only afforded 8% of the oxazoline product 3 (Table 1, entry 13). These reactions validated the critical roles of each individual component to achieve an efficient reaction.
To understand this coupling reaction better, we have also performed time studies to elucidate the effects of several key features in this reaction. First, we studied the iodoarene catalyst precursor and the lithium salt in terms of their effects on the overall reaction rate. In this case, we observed that the overall reaction proceeded faster with the more electron-rich iodoarene catalysts than electron-poor ones. Qualitatively, the electron-rich iodoarene catalysts are likely to be worse at activating the olefins than the electron-deficient hypervalent iodine catalysts. Therefore, the faster rate with the electron-rich catalyst precursor is because the electron-rich iodoarene catalyst precursors are more easily oxidized to the hypervalent iodine catalyst with difluoro ligands. Interestingly, the use of different lithium salts also impacted the overall reaction rate, with the reaction using the less coordinating LiAsF_6_ salt proceeding faster than LiPF_6_ and LiBF_4_. This time study suggested that the hypervalent iodine precatalyst with the less coordinating counterion is more reactive to activate the olefin. We also conducted kinetic studies on how the olefin and amide structures impacted the overall reaction rate. The more electron-rich olefins generally proceeded faster than the electron-poor ones, suggesting that a significant positive charge was likely built up on the olefin prior to the nucleophilic addition. On the other hand, the electronic nature of the para-substituted benzamides had little impact on the overall reaction rate as both electron-rich and electron-deficient benzamides proceeded with similar kinetic profiles. All the kinetic plots are shown in Figure 1.
Time studies of the amide and alkene coupling. a) Iodoarene time studies: styrene (1), para-substituted iodoarenes, LiBF4, and benzamide (2). b) Li salt time studies: styrene (1), iodoanisole, Li salts, and benzamide (2). c) Alkene time studies: para-substituted styrenes, iodotoluene A, LiBF4, and benzamide (2). d) Benzamide time studies: styrene (1), iodotoluene A, LiBF4, and para-substituted benzamides.
With the optimized conditions and kinetic information in hand, we turned our attention to the amide substrate scope. In this case, both electron-rich and -deficient benzamides proceeded to the desired products in reasonable yields and high regioselectivities (Figure 2, products 4–7). Concurring with our kinetic data, the electronic nature of the amide bears little impact on the overall reaction rate and in this case, on the final yields as well. Similarly, ortho- and meta-substituted benzamides with halogen functionalities could also generate the desired oxazoline products with reasonable yields (Figure 2, products 8–10). Heteroaromatic amides could also furnish the oxazolines 11 and 12 with good efficiency. Naphthaleneamide also generated the desired product 13, albeit with slightly lower efficiency. Interestingly, o- and m-methyl-substituted benzamides provided a significant yield boost to provide the oxazoline structures 14 and 15. Finally, sterically encumbered tertiary amides participated in the reaction to afford the respective regioisomeric product 16.
Amide substrate scope studies. a) Standard conditions: styrene (0.25 mmol), iodotoluene (20 mol %), LiBF4 (100 mol %), Selectfluor (150 mol %), amide (400 mol %), MeNO2 (0.25 M), rt, 16 h. b) Iodoanisole (20 mol %).
Encouraged by these results, we then turned our attention to explore the extent of alkene substrate scope using 3,4-dimethylbenzamide, which afforded the oxazoline product 17 in 70% yield (Figure 3). Based on this optimal amide structure, we examined various electronically activated olefins under the optimal reaction conditions. A number of styrenyl derivatives with para-substituted halogens, ester, and phthalimide proceeded smoothly with good yields and excellent regioselectivities to access the oxazoline products as single regioisomers (Figure 3, products 18–22). The o-bromo-substituted styrene also afforded the corresponding product 23. Furthermore, 1,1-di-substituted α-methylstyrene and α-phenylstyrene produced the respective oxazoline products with high regioselectivity and reasonable yields using iodoanisole as the catalyst precursor (Figure 3, products 24 and 25).
Alkene substrate scope studies. a) Standard conditions: alkene (0.25 mmol), iodotoluene (20 mol %), LiBF4 (100 mol %), Selectfluor (150 mol %), 3,4-dimethylbenzamide (400 mol %), MeNO2 (0.25 M), rt, 16 h. b) Iodoanisole (20 mol %), MeCN (0.25 M).
The proposed catalytic cycle (Figure 4) begins with iodotoluene A which is oxidized by Selectfluor salt into the difluorinated iodotoluene B. Then, LiBF_4_ can perform a salt metathesis with B to produce LiF along with the active hypervalent iodoarene catalyst C. The activated hypervalent iodine catalyst C can coordinate to the alkene to form complex D. The nucleophilic oxygen of the amide will attack in the internal position and subsequent cyclization will furnish the desired oxazoline.
Proposed catalytic cycle for the hypervalent iodine-catalyzed amide and alkene coupling.
Conclusion
We have developed a hypervalent iodine-catalyzed amide and alkene coupling reaction. This reaction protocol furnished useful oxazoline products and introduced the use of lithium salts to activate hypervalent iodine catalysts. This strategy rendered the participation of simple and unadorned amides as bifunctional nucleophiles to achieve olefin oxyamination reactions. Time studies of these reactions further unveiled interesting mechanistic features that will be useful for our future catalysis development and asymmetric reaction designs.
Supporting Information
File 1Spectral characterization of the products and kinetic studies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Zhdankin V V Stang P J Chem Rev 20021022523258410.1021/cr 010003+12105935 · doi ↗ · pubmed ↗
- 2Zhdankin V V Stang P J Chem Rev 20081085299535810.1021/cr 800332 c 18986207 PMC 2736367 · doi ↗ · pubmed ↗
- 3Wirth T Top Curr Chem 200322414
- 4Merritt E A Olofsson B Angew Chem, Int Ed 2009489052907010.1002/anie.20090468919876992 · doi ↗ · pubmed ↗
- 5Yoshimura A Zhdankin V V Chem Rev 20161163328343510.1021/acs.chemrev.5b 0054726861673 · doi ↗ · pubmed ↗
- 6Ochiai M Miyamoto K Eur J Org Chem 20084229423910.1002/ejoc.200800416 · doi ↗
- 7Dohi T Kita Y Chem Commun 20092073208510.1039/b 821747 e 19360157 · doi ↗ · pubmed ↗
- 8Fuchigami T Fujita T J Org Chem 1994597190719210.1021/jo 00103 a 003 · doi ↗