TGFβ signalling: a nexus between inflammation, placental health and preeclampsia throughout pregnancy
Monika Horvat Mercnik, Carolin Schliefsteiner, Gonzalo Sanchez-Duffhues, Christian Wadsack

TL;DR
This review explores how TGFβ signaling regulates placental development and immune tolerance during pregnancy, and how its disruption may lead to preeclampsia.
Contribution
The paper provides a comprehensive overview of TGFβ superfamily members' roles in placental cell functions and their dysregulation in preeclampsia.
Findings
TGFβ signaling regulates trophoblast invasion and epithelial-to-mesenchymal transition during early pregnancy.
Dysregulation of TGFβ signaling is linked to preeclampsia, affecting vascularization and immune tolerance.
TGFβ signaling influences placental macrophage polarization and maternal tolerance of the fetus.
Abstract
The placenta is a unique and pivotal organ in reproduction, controlling crucial growth and cell differentiation processes that ensure a successful pregnancy. Placental development is a tightly regulated and dynamic process, in which the transforming growth factor beta (TGFβ) superfamily plays a central role. This family of pleiotropic growth factors is heavily involved in regulating various aspects of reproductive biology, particularly in trophoblast differentiation during the first trimester of pregnancy. TGFβ signalling precisely regulates trophoblast invasion and the cell transition from cytotrophoblasts to extravillous trophoblasts, which is an epithelial-to-mesenchymal transition-like process. Later in pregnancy, TGFβ signalling ensures proper vascularization and angiogenesis in placental endothelial cells. Beyond its role in trophoblasts and endothelial cells, TGFβ signalling…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
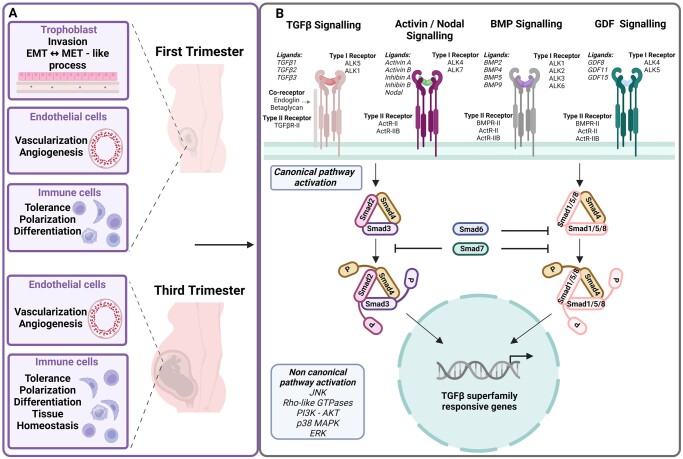 Figure 1
Figure 1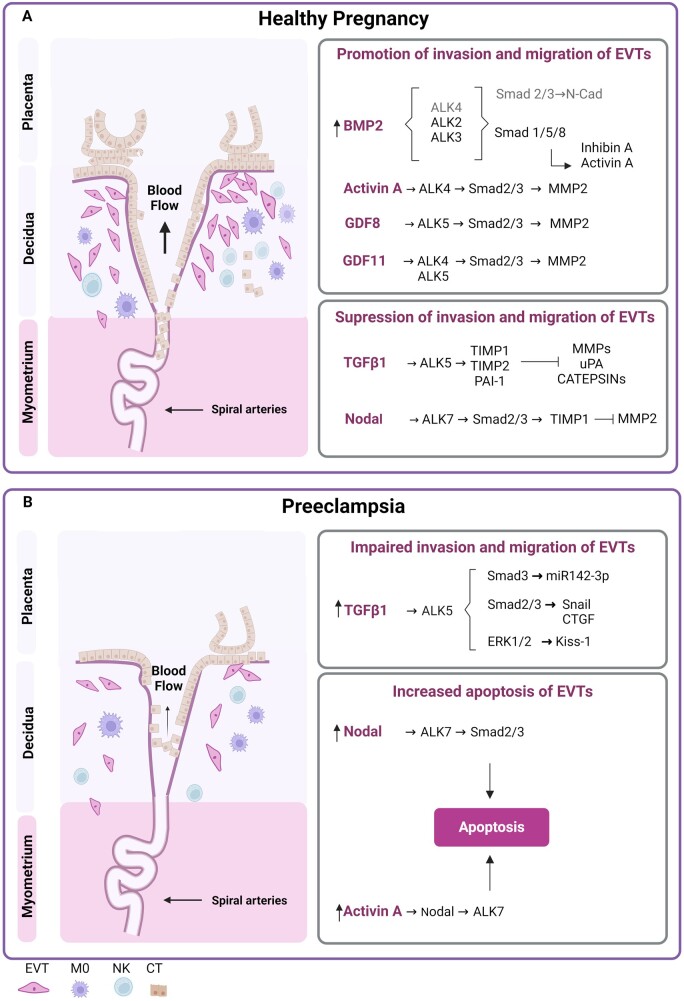 Figure 2
Figure 2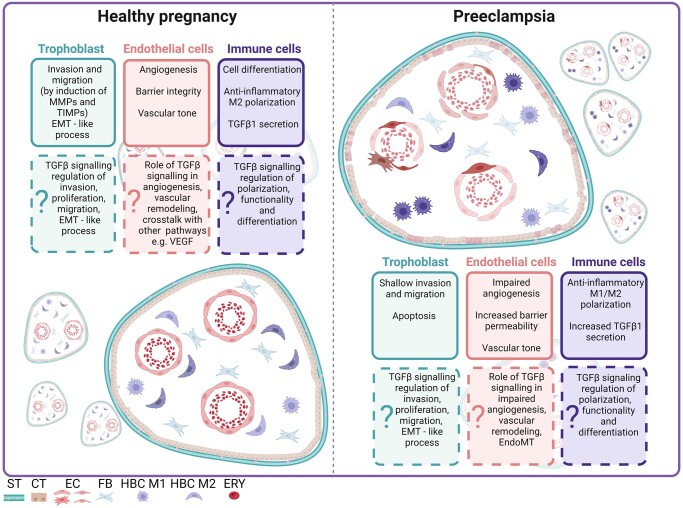 Figure 3
Figure 3| TGFβ superfamily | Ligand | Compartment | Tissue/cell type | Reference |
|---|---|---|---|---|
| TGFβ | TGFβ1 | Placenta | CT |
|
| EVT |
| |||
| ST |
| |||
| dNK |
| |||
| dM |
| |||
| HBC |
| |||
| Maternal tissue | Decidua, Endometrium, Uterus |
| ||
| TGFβ2 | Placenta | CT |
| |
| EVT |
| |||
| dM |
| |||
| HBC |
| |||
| Maternal tissue | Decidua, Endometrium, Uterus |
| ||
| TGFβ3 | Placenta | EVT |
| |
| HBC |
| |||
| Maternal tissue | Decidua, Endometrium, Uterus |
| ||
| BMP | BMP2 | Placenta | EVT |
|
| HBC |
| |||
| Maternal tissue | Decidua |
| ||
| BMP4 | Foetal tissue | Extraembryonic ectoderm |
| |
| BMP9 | Maternal tissue | Liver |
| |
| NODAL | NODAL | Maternal tissue | Uterus |
|
| ACTIVIN/INHIBIN | ACTIVIN A | Placenta | CT |
|
| Maternal tissue | Decidua |
| ||
| Foetal tissue | Foetal membranes |
| ||
| ACTIVIN B | Placenta | CT |
| |
| INHIBIN A | Placenta | CT |
| |
| Maternal tissue | Decidua |
| ||
| Foetal tissue | Foetal membranes |
| ||
| INHIBIN B | Placenta | CT |
| |
| ST |
| |||
| Maternal tissue | Decidua |
| ||
| Foetal tissue | Foetal membranes |
| ||
| GDF | GDF8 | Placenta | CT |
|
| ST |
| |||
| EVT |
| |||
| Maternal tissue | Uterus, Endometrium |
| ||
| GDF11 | Placenta | CT |
| |
| ST |
| |||
| GDF15 | Placenta | CT |
| |
| ST |
| |||
| EVT |
| |||
| Maternal tissues | Decidua |
|
| TGFβ superfamily ligand | Subtype of PE | Type of control | Sample | Changes of ligand levels in PE | Reference |
|---|---|---|---|---|---|
| TGFβ1 | Mild/Severe EO/LOPE |
Normotensive pregnancy (Unmatched for GA) | Plasma | Elevated in all PE subtypes |
|
| PE | Normotensive pregnancy (Matched for GA) | Plasma | Elevated |
| |
| Mild/Severe/PE+ FGR | Normotensive pregnancy (Unmatched for GA) | Plasma | Active TGFβ elevated in all PE subtypes |
| |
| PE | Normotensive pregnancy (Matched for GA) | Serum | No difference in total or active TGFβ1 |
| |
| PE | Normotensive pregnancy (Unmatched for GA) | Serum | No difference in total or active TGFβ1 |
| |
| Mild/Severe PE |
Normotensive pregnancy (Matched for GA) | Serum | Decreased levels in mild PE, elevated in severe PE |
| |
| PE | Normotensive pregnancy (Matched for GA) | Placenta | Elevated |
| |
| PE | Normotensive pregnancy (Unmatched for GA) | Placenta | Elevated |
| |
| PE | Preterm normotensive pregnancy | Placenta | Elevated |
| |
| PE | Normotensive pregnancy (Unmatched for GA) | Placenta | No difference |
| |
| TGFβ2 | Mild/Severe PE/Eclampsia | Normotensive pregnancy (Matched for GA) | Serum | Elevated in Severe PE and Eclampsia |
|
| TGFβ3 | PE | Normotensive pregnancy (Matched for GA) | Placenta | Elevated |
|
| EOPE | Preterm normotensive pregnancy | Placenta | Reduced |
| |
| BMP2 | EOPE | Preterm normotensive pregnancy | Placenta | Reduced |
|
| BMP4 | PE |
Normotensive pregnancy (Unmatched for GA) | Serum | No difference |
|
| EOPE | Preterm normotensive pregnancy | Placenta | Reduced |
| |
| BMP5 | EOPE | Preterm normotensive pregnancy | Placenta | Reduced |
|
| Nodal | PE |
Preterm normotensive pregnancy (Matched for GA) | Placenta | Elevated |
|
| Activin A | Mild/Severe PE/Gestational hypertension |
Normotensive pregnancy (Matched for GA) | Serum | Elevated in severe PE > mild PE and gestational hypertension |
|
| PE |
Normotensive pregnancy (Matched for GA) | Serum | Elevated |
| |
| PE |
Normotensive pregnancy (Unmatched for GA) | Serum | Elevated |
| |
| PE |
Normotensive pregnancy (Unmatched for GA) | Placenta | Elevated |
| |
| Inhibin A | Mild/Severe PE/Gestational hypertension |
Normotensive pregnancy (Matched for GA) | Serum | Elevated in severe PE > mild PE and gestational hypertension |
|
| PE |
Normotensive pregnancy (Matched for GA) | Serum | Elevated |
| |
| EOPE | Preterm normotensive pregnancy | Placenta | Elevated |
| |
| Inhibin B | PE |
Normotensive pregnancy (Matched for GA) | Serum | No difference |
|
| EOPE | Preterm normotensive pregnancy | Placenta | Elevated |
| |
| Myostatin (GDF8) | Mild/Severe PE |
Normotensive pregnancy (Matched for GA) | Serum | Elevated |
|
| Mild/Severe PE |
Normotensive pregnancy (Matched for GA) | Placenta | Elevated |
| |
| Endoglin | Mild/Severe PE |
Normotensive pregnancy (Unmatched for GA) | Plasma | Increasing with the severity of PE |
|
| Mild/Severe PE |
Normotensive pregnancy (Matched for GA) | Serum | Increasing with the severity of PE |
| |
| Mild/Severe PE |
Normotensive pregnancy (Unmatched for GA) | Serum | Increasing with the severity of PE |
| |
| Mild/Severe PE |
Normotensive pregnancy (Matched for GA) | Placenta | Increasing with the severity of PE |
| |
| Follistatin related gene protein | PE |
Normotensive pregnancy (Matched for GA) | Serum | Elevated |
|
| PE |
Normotensive pregnancy (Matched for GA) | Placenta | Elevated |
| |
| FSTL3 | Mild/Severe PE |
Normotensive pregnancy (Matched for GA) | Serum | Elevated |
|
| Mild/Severe PE |
Normotensive pregnancy (Matched for GA) | Placenta | Elevated |
|
| TGFβ superfamily | Ligand | Model system | Studied trophoblast function | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| TGFβ | TGFβ1 | Primary trophoblast | Differentiation of placental EVTs into intestitial EVTs |
| |||||||
| TGFβ targets expression |
| ||||||||||
| Invasion |
| ||||||||||
| Placental explants | Invasion |
| |||||||||
| Cell lines | JEG-3 | Invasion |
| ||||||||
| HTR8/SVneo | Invasion |
| |||||||||
| Organoids | Differentiation of placental EVTs into intestitial EVTs |
| |||||||||
| TGFβ3 | Cell lines | JAR | Invasion |
| |||||||
| JEG-3 | Invasion |
| |||||||||
| Placental explants | Invasion |
| |||||||||
| BMP | BMP2 | Primary trophoblasts | Invasion |
| |||||||
| Differentiation |
| ||||||||||
| Cell lines | HTR8/SVneo | Invasion |
| ||||||||
| Endothelial-like tube formation |
| ||||||||||
| BMP4 | Trophoblast stem cells (TSC) | Derivation of TSCs from pluripotent stem cells |
| ||||||||
| Nodal | Nodal | Primary trophoblasts | Apoptosis |
| |||||||
| Placental explants | Invasion |
| |||||||||
| Proliferation |
| ||||||||||
| Cell lines | JAR | Proliferation |
| ||||||||
| JEG-3 | Proliferation |
| |||||||||
| Bewo | Proliferation |
| |||||||||
| HTR8/SVneo | Invasion |
| |||||||||
| Proliferation |
| ||||||||||
| Apoptosis |
| ||||||||||
| Trophoblast stem cells | EVT formation |
| |||||||||
| Activin/Inhibin | Activin A | Primary trophoblasts | Differentiation |
| |||||||
| Invasion |
| ||||||||||
| Apoptosis |
| ||||||||||
| Hormone production |
| ||||||||||
| Placental explants | Differentiation |
| |||||||||
| EVT formation |
| ||||||||||
| Invasion |
| ||||||||||
| Cell lines | HTR8/SVneo | Invasion |
| ||||||||
| Apoptosis |
| ||||||||||
| Inhibin A | Placental explants | EVT growth |
| ||||||||
| GDF | GDF8 | Primary trophoblast | Invasion |
| |||||||
| Cell lines | HTR8/SVneo | Invasion |
| ||||||||
| GDF11 | Primary trophoblast | Invasion |
| ||||||||
| Cell lines | HTR8/SVneo | Invasion |
| ||||||||
| GDF15 | Placental Explants | Ligand expression |
| ||||||||
| Cell lines | HTR8/SVneo | Ligand expression |
| ||||||||
| Apoptosis |
| ||||||||||
| Proliferation |
| ||||||||||
| Model | Origin | Strengths | Limitations | References |
|---|---|---|---|---|
| Primary trophoblasts | First and third trimester placenta |
Preserve original genetic characteristics Closely resemble the Accessible samples obtained from term healthy and pathophysiological pregnancies Well established isolation, characterization, and cryopreservation protocols. |
Limited proliferation ability Limited self-renewal Mixed cell populations (CTs, STs…) Reduced invasiveness and motility of EVTs at term Difficult to acquire second trimester placental samples Lack of physiological microenvironment and other cell types |
|
| Placental explants | First and third trimester placenta |
Retain the Study cell-to-cell interactions and responses Investigate cellular metabolism under healthy and pathophysiological conditions Flow-cultured explants preserve the tissue intactness |
Shorter cultivation time in the static settings Limited self-renewal Genetic manipulation |
|
| Cell lines | Derived from:
Choriocarcinoma: JAR JEG-3 Bewo Primary first trimester extravillous trophoblasts transfected with simian virus 40 large T-antigen (SV40T): HTR8/SVneo |
Display (some) properties of EVTs Easy to maintain Genetic manipulation Stable and reproducible Time-dependent experimental settings allow dynamic studies of placental development Used to study invasion, proliferation and regulation |
Immortalized cells Genetic alterations and phenotypical differences as primary trophoblasts Mimic specific phenotypes of EVTs or STs Limited functional differentiation Cell lines observed signalling do not completely manifest the pathway signalling in primary trophoblasts |
|
| Trophoblast stem cells (TSC) | Derived from:
Human placental trophoblast tissue Human pluripotent stem cells (PSCs)/human embryonic stem cells (ESCs)/human-induced pluripotent stem cells (iPSCs) and human ePSCs stimulated with inducing factors as BMP4 |
Normal karyotype Unlimited proliferation Multi-directional differentiation Similar gene expression as primary trophoblasts Studies on pathophysiological cells as in PE Easy genetic manipulation Cell fusion/differentiation is inducible |
Genetic heterogeneity Widespread imprint erasure High demanding complex culturing process Higher costs |
|
| Trophoblast organoids | First trimester primary trophoblasts/JEG-3/third trimester trophoblasts/TSC |
Structural and transcriptional similarity to placental villi, allowing biological dynamics to mimic developmental processes Capable of proliferation and self-renewal, fusion and secretion of placental hormones Long-term culturing and cryopreservation |
Organoids represent expression profile of first trimester placental tissue, therefore might not be suitable as a model for third trimester placenta Combination of primary cells and immortalized cell lines Complex culturing system Costly |
|
| Placenta-on-chip |
Primary trophoblast/ JEG-3, Co-culture with endothelial cells, fibroblasts |
Microfluidics system Control of culture microenvironment Real-time trophoblast differentiation Study of placental barrier, metabolism, transport, and cell-to-cell interactions |
Combination of primary cells and immortalized cell lines Costly |
|
| Animal models | Rodents, non-human primates |
Study TGFβ signalling in the context of tissue microenvironment Investigate |
Anatomical and physiological differences between species The structural and functional differences in placental anatomy across pregnancy Genetic manipulation Mimic pathophysiological conditions as PE Costly Ethics |
|
- —Program Inflammatory Disorders in Pregnancy
- —Medical University of Graz, Austria
- —Austrian Science Fund FWF
- —Fundació La Marató de TV310.13039/100008666
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
Topicsnanoparticles nucleation surface interactions · Fluid Dynamics and Heat Transfer · Heat Transfer and Boiling Studies
Introduction
During pregnancy, the human placenta plays a unique role as the first organ to develop from cells originating from the blastocyst. The beginning of a healthy pregnancy relies on the intricate regulation between trophoblast cell invasion and foeto-maternal tolerance (Knöfler et al., 2019). As the pregnancy advances, the successful maintenance of the pregnancy relies on the precise coordination, action, and interplay of various cell types in a highly orchestrated manner, to support the developing foetus and ensure a healthy pregnancy outcome (Huppertz, 2008a; Cindrova-Davies and Sferruzzi-Perri, 2022). TGFβ signalling plays a major part in embryonic and placental development and thereby effecting pregnancy outcome (Fig. 1) (Haider et al., 2017, 2022; Li et al., 2021; Yang et al., 2021; Fang et al., 2022). In the first trimester of pregnancy, TGFβ signalling is primarily involved in the regulation of trophoblast invasion, remodelling of the spiral arteries, and development of the placental vasculature (Dietrich et al., 2022; Li et al., 2023a). Disturbances in TGFβ levels in maternal plasma have been associated with miscarriages (Ogasawara et al., 2000; Dirisipam et al., 2023), but TGFβ levels in the placental bed do not seem to be altered under these circumstances (Ball et al., 2007). Emerging evidence suggest that various components of the TGFβ pathways have been frequently reported as altered in the pathogenesis of preeclampsia (PE) (Venkatesha et al., 2006; Haider et al., 2017; Zhao et al., 2020b; Deng et al., 2023), and might be potential therapeutic targets for intervention. As the pregnancy progresses, TGFβs take up the role as immune suppressors, promoting maternal tolerance of the semi-allogenic allograft during gestation (Ingman and Robertson, 2009). Additionally, TGFβ family members are involved in regulating the function of placental endothelial cells (ECs) (Zhou et al., 2019) and immune cells (Keskin et al., 2007; Mercnik et al., 2022; Vondra et al., 2023). This comprehensive review highlights the TGFβ superfamily signalling and its key ligands, such as transforming growth factor β (TGFβ), bone morphogenetic proteins (BMPs), activins, inhibins, nodals, and growth differentiation factors (GDFs), in the context of placental function in normal and PE-compromised pregnancies. In consequence, all major cell types of the early and late placenta namely, trophoblasts, ECs, and immune cells, are the subject of these summaries. By highlighting the indispensable role of TGFβ family members, this review emphasizes the inherent biological diversity of placental cells and related cell models, which are, in addition, influenced by non-placental cell types that produce vital TGFβ ligands. While placental cells are the primary source of TGFβ ligands (Jones et al., 2006b; Chuva de Sousa Lopes et al., 2020), it is also crucial to note the involvement of other non-placental factors. For example, the release of TGFβ ligands from foetal or maternal tissues, that is the liver (Bidart et al., 2012) or the endometrium (Jones et al., 2006b), thereby, contributing to the complex network of TGFβ signalling in the human placenta (Table 1). In detail, this substantial contribution of TGFβ family ligands from maternal and foetal cell types beyond the placenta, influencing the maternal–placental interface, is essential for successful pregnancy initiation, and involves cellular interactions and networks, thereby intricately shaping placental function and development. Accounting for ligand bioavailability broadens the signalling window through the TGFβ receptor family, adding complexity to the understanding of the TGFβ cellular network and highlighting the critical importance of TGFβ signalling in the context of pregnancy.
TGFβ signalling in the placenta. (A) TGFβ signalling in the first and third trimester placenta. TGFβ signalling is crucial for placental development and the functional regulation of placental cells. In trophoblasts, particularly during the first trimester, TGFβ signalling is involved in regulating cell invasion and the transition of CTs to EVTs through an EMT-like process. Additionally, some ligands of the TGFβ family stimulate the reverse process called MET. To ensure blood supply and placental function, TGFβ signalling contributes to vascularization by stimulating angiogenesis through placental ECs. During pregnancy, TGFβ signalling contributes to the establishment of the maternal tolerance to the semi-allogeneic foetus by limiting the immunogenic response. TGFβ also plays a central role in Tregs differentiation, regulation of NK cells functions, and the balance between M1 and M2 macrophages. (B) TGFβ family signalling activation pathways in the human placenta. TGFβ ligands interact with type I and type II receptors and induce downstream signalling through canonical and non-canonical pathways. Upon ligand-receptor binding, the receptor complex becomes phosphorylated and recruits the canonical intracellular mediators Smad2/3 or Smad1/5/8. Depending on the type I receptor activated, in the canonical signalling route, either Smad2/3 or Smad1/5/8 are trans-phosphorylated. TGFβ and Activins via ALK4/5/7 primarily activate Smad2/3, whereas Smad1/5/8 activation is induced by the BMP receptors ALK1/2/3/6. Of note, on certain occasions, TGFβ s, activins, and GDFs can activate both Smad1/5/8 and Smad2/3 signalling. Phosphorylated Smads form complexes with Smad4 and translocate to the nucleus to regulate gene expression. Inhibitory Smads, such as Smad6 and Smad7, act as negative feedback regulators to balance receptor activity. In addition to the canonical pathways, TGFβ ligand binding can activate non-canonical, Smad-independent signalling pathways. These pathways involve downstream effectors such as JNK, Rho-like GTPases, PI3K-AKT, p38 MAPK, and ERK. Non-canonical signalling regulates TGFβ responsive genes and mediates additional cellular responses.
Transforming growth factor beta signalling
Transforming growth factor beta (TGFβ) signalling is vital in tissue and cell homeostasis, regulating inflammatory and cellular processes such as proliferation, growth, differentiation, apoptosis, migration, or matrix formation (Moustakas et al., 2002; Tzavlaki and Moustakas, 2020). Since the discovery of the first ligand namely TGFβ1 (Assoian et al., 1983), more than thirty ligands have been described (Derynck and Budi, 2019). The characterization of TGFβ ligands has been based on structure and phylogeny (Heldin and Moustakas, 2016), sub-stratifying them into six major subfamilies: the TGFβ isoforms, BMPs, nodals, anti-Müllerian hormone (AMH), activins/inhibins, and GDFs (Derynck and Budi, 2019). This review focuses on the TGFβ subfamilies and their roles in healthy and preeclamptic placentas (Fig. 1). While AMH is vital for the ovarian reserve and for detecting polycystic ovary syndrome (Rudnicka et al., 2021), its role in placental development remains unclear. Although AMH and its receptor are present in placental tissue (Novembri et al., 2015), their direct significance and function in this context are yet to be understood, and therefore, this review will not delve into it.
All members of the TGFβ family act as homo- or heterodimers, in most cases formed through a disulfide bond (Heldin and Moustakas, 2016). Activation of the TGFβ superfamily begins with ligand dimer binding to transmembrane serine–threonine kinase receptors, which are divided into type I and type II classes. There are seven type I receptors, known as activin receptor-like kinases (ALK1–ALK7) and five type II receptors: activin type II receptor A and B (ActRIIA, ActRIIB), BMP receptor II (BMPRII), transforming growth factor β receptor II (TGFβRII), and anti-Müllerian hormone receptor II (AMHRII) (Sierra-Filardi et al., 2011; Heldin and Moustakas, 2016; Derynck and Budi, 2019; Tzavlaki and Moustakas, 2020). Co-receptors such as endoglin (Eng), BMP and activin membrane-bound inhibitor (BAMBI), betaglycan (TGFΒRIII), cripto, CD109, repulsive guidance molecule b (RGMb), and neuropilin-1 further affect the signalling (Vogt et al., 2011; Tzavlaki and Moustakas, 2020).
Canonical Smad pathway
TGFβ family members exhibit the conventional activation of canonical Smad (derived from the fusion of the Caenorhabditis elegans Sma genes and the Drosophila Mad—Mothers against decapentaplegic—genes) mediated pathways (Moustakas and Heldin, 2009; Derynck and Budi, 2019; Tzavlaki and Moustakas, 2020). The Smad family consist of eight Smad proteins, which are divided into three distinct subgroups: receptor-activated Smads (R-Smads: Smad1, Smad2, Smad3, Smad5, and Smad8), the common Smad (C-Smad4), and inhibitory Smads (I-Smads: Smad6 and Smad7) (Heldin et al., 1997; Wrighton et al., 2009).
To initiate the signalling cascade, a dimeric TGFβ ligand interacts with the specific type I and type II receptors to form a heterotetrameric complex. Upon a complex formation, type II receptors activate the type I receptor by trans-phosphorylation (Heldin and Moustakas, 2016). This phosphorylation occurs in the juxtamembrane domain of the type I receptor and activates its kinase function (Moustakas and Heldin, 2009; Heldin and Moustakas, 2016; Yan et al., 2018). Followed by a successful receptor activation, the R-Smads are recruited and activated by the type I receptor to form cytosolic heteromeric trimers together with Smad4 (Tzavlaki and Moustakas, 2020). Smad4 functions as a cofactor that enhances ligand-induced transcription by stabilizing the interaction of R-Smads with DNA (Hill, 2016; Kamato et al., 2020). Although Smad4 is not essential for nuclear transport, it is often translocated together with R-Smads. In the nucleus, Smads form transcriptional complexes with specific cofactors and regulatory proteins, thereby influencing the expression of target genes (Miyazono et al., 2005). TGFβ ligands can activate both the Smad2/3 and Smad1/5/8 pathways in a context-dependent manner. In particular, Smad1, Smad5, and Smad8 are primarily R-Smads activated by BMP type I receptors, whereas Smad2 and Smad3 are known to be activated by activin, GDF, and TGFβ type I receptors (Rochette et al., 2020). However, it is important to emphasize that activation of the Smad2/3 or the Smad1/5/8 pathway is context dependent and varies according to ligand binding, receptor activation, and conditions (Heldin and Moustakas, 2016; Rochette et al., 2020; Tzavlaki and Moustakas, 2020).
Post-translational modifications of Smads fine-tune the responses to TGFβ ligands. Phosphorylation, ubiquitination, sumoylation, and other modifications occur to control Smad stability and function. In the nucleus, R-Smads are constantly dephosphorylated, leading to dissociation of Smad complexes and the export of inactive Smads to the cytoplasm (Derynck and Zhang, 2003; Vogt et al., 2011). Moreover, Smad signalling is regulated by positive and negative regulators, including I-Smads, the co-repressors Ski and SnoN and the Smurf family of E3 ubiquitin ligases (Luo, 2017). The I-Smads are strongly induced by TGFβ receptor activation, and their induction provides an auto-inhibitory feedback mechanism for ligand-induced signalling (Yan et al., 2016). Smad6 and Smad7 compete with R-Smads for receptor interaction. They can also induce receptor degradation or interfere with Smad DNA binding (Yan et al., 2009, 2016). In addition, Smads are involved in crosstalk with other signalling pathways, such as the Wingless-related integration site (Wnt), Hippo, Notch, Hedgehog, and Nuclear Factor κB (NF-κB) pathways, often in a cell type-specific manner, which plays an important role in the regulation of various biological responses (Luo, 2017).
Non-canonical pathway
In addition to canonical Smad signalling, TGFβ-related ligands can also regulate other signalling pathways, that may influence both Smad-mediated and Smad-independent responses (Vogt et al., 2011). Smad-independent signalling or non-canonical TGFβ signalling involves the activation of several downstream pathways, including: c-JUN N-terminal kinase (JNK), Rho-like GTPases, phosphatidylinositol 3-kinase (PI3K)—protein kinase B (Akt), p38 mitogen-activated kinase (MAPK), and extracellular signal-regulated kinase (ERK) MAPK (Luo, 2017; van Caam et al., 2017). The MAPK and Smad pathways often directly or reciprocally regulate each other’s activities and output. For example, ERK and GSK3b kinases can phosphorylate the R-Smads in the linker region, to either enhance or diminish Smad mediated transcriptional activation (Fuentealba et al., 2007; Hough et al., 2012). Moreover, activation of non-canonical signalling leads to the differential recruitment of cofactors to transcriptional complexes containing the Smads, thereby influencing the selectivity, potency, and longevity of Smads-induced gene expression (Abdollah et al., 1997; Derynck and Budi, 2019). TGFβRI mediated phosphorylation of TAK1 (TGFβ activated kinase 1), which is an important mediator of p38 MAPK pathway, has been implicated in the regulation of apoptosis, cell migration, and cell differentiation (Wendt et al., 2009; Derada Troletti et al., 2019). TGFβ family ligands induce a rapid activation of Rho-like GTPases resulting in control of cytoskeletal dynamics and cell motility (Derynck and Zhang, 2003; Derynck and Budi, 2019). Moreover, TGFβ can indirectly activate PI3K-Akt signalling through miRNAs (Zhang, 2017). In turn, inhibition of PI3K-Akt signalling has been shown to prevent TGFβ-induced Smad3 activation and the association with Smad4 (Runyan et al., 2004). PI3K-Akt signalling has been shown to antagonize TGFβ/Smad signalling to promote cell survival through an Akt kinase-dependent mechanism (Zhang, 2017).
TGFβ superfamily ligands
TGFβ subfamily
TGFβ exists in three known isoforms: TGFβ1, TGFβ2, and TGFβ3, all of which are secreted into the extracellular space in an inactive, latent form (Jenkins, 2008). The TGFβ isoforms contain highly conserved regions but differ in several amino acid sequences (Kubiczkova et al., 2012; Poniatowski et al., 2015). Despite sequence similarities, the isoforms bind differently to the receptors. TGFβ2 binds to the TGFβRII through different residues than TGFβ1 and TGFβ3, and presentation of TGFβ2 to the receptor requires the presence of a co-receptor, either betaglycan or Eng (Heldin and Moustakas, 2016), whereas TGFβ1 and TGFβ3 can bind directly to TGFβRII (Laverty et al., 2009; Huang et al., 2014). Although the isoforms signal via similar signalling pathways, the cellular effect and outcome may differ. TGFβ1 is the most abundant and ubiquitously expressed isoform, and is predominantly expressed in the cells of the immune system, where it acts as a potent immunoregulator (Yoshimura et al., 2010; Kubiczkova et al., 2012). Activation of latent TGFβ is an important checkpoint of TGFβ bioavailability (Jenkins, 2008). To prevent access to TGFβ receptors, ligands are synthesized as precursors (18) consisting of a pro-domain, called latency associated polypeptide (LAP) and the active TGFβ. The precursors can be cleaved into mature dimeric proteins, often linked by disulfide bonds during processing through the secretory pathway. Proteases play a role in the process of indirect or direct activation of extracellular TGFβ. Some matrix metalloproteinases (MMPs), such as MMP2 and MMP9 can directly cleave and activate latent TGFβ, whereas membrane-type matrix metalloproteinase (MT-MMP) interacts with integrin-mediated TGFβ activation pathways (Jenkins, 2008). Active TGFβ is involved in the synthesis of many of its own activators, such as thrombospodin-1 (TSP-1), furins, and metalloproteinases (De Caestecker et al., 1998), creating autocrine feedback loops in TGFβ bioavailability and signalling.
Upon activation of the ligands, they bind to their respective receptors. TGFΒRII is primarily involved in the basic signal transduction of the TGFβ subfamily as a type II receptor (Moustakas and Heldin, 2009; Heldin and Moustakas, 2016). In most cell types, TGFβ signals via ALK5 or TGFβRI as a type I receptor, whereas in ECs signal transduction can occur via both ALK5 and ALK1 (Goumans et al., 2002, 2003). Signalling via ALK5 activates Smad2/3 signalling (Tzavlaki and Moustakas, 2020), which is considered the most canonical TGFβ signalling, while stimulation of ALK1 leads to activation of Smad1/5/8 (Goumans et al., 2003). The coreceptor Eng is required for TGFβ signalling via ALK1. Other coreceptors, like betaglycan or TGFΒRIII, may also regulate signalling by either facilitating or inhibiting ligand–receptor interaction, adding another layer of regulation and specificity to the signalling cascade (Heldin et al., 1997; Moustakas and Heldin, 2009; Heldin and Moustakas, 2016; Tzavlaki and Moustakas, 2020).
BMP subfamily
Since their discovery as ectopic bone inducers, BMPs have been shown to affect a wide variety of cell types and processes beyond bone physiology (Wang et al., 2014). They are important morphogens in embryogenesis and development and have also been shown to regulate adult tissue homeostasis (Wang et al., 2014). Many processes in early development depend on BMP signalling gradients for cell growth, apoptosis, and differentiation (Zou and Niswander, 1996; Kobayashi et al., 2005; Jones et al., 2006b; Stewart et al., 2010; Wang et al., 2014). Based on the sequence similarity and their function, BMPs are divided into several subgroups: (i) BMP2/4, (ii) BMP5/6/7/8a/8b, (iii) BMP9/10, and (iv) BMP12/13/14 (Bragdon et al., 2011). Normally, BMPs are synthesized as pro-peptides, and after secretion and cleavage, BMPs bind to either the extracellular matrix, soluble antagonists, co-receptors, or transmembrane serine/threonine kinase receptors (Sieber et al., 2009). Similar to TGFβ, BMPs bind to heteromeric receptor complexes composed of type I and type II (BMPRII, ActRII, ActRIIB) transmembrane serine/threonine kinase receptors and activate the corresponding signalling pathway (Von Bubnoff and Cho, 2001; Miyazono et al., 2005; Xiao et al., 2007; Sieber et al., 2009). In general, BMPs are thought to bind to the type I receptors ALK1/2/3/6 and activate the Smad1/5/8 pathway (Tzavlaki and Moustakas, 2020).
Nodal subfamily
Nodal is a multifunctional cytokine that plays a central role in the embryonic stages of mammals and is important in regulating placental development. It does this by suppressing trophoblast proliferation and inhibiting both extravillous trophoblast (EVT) formation and invasion of the decidua (Sarkar et al., 2015). Nodal binds to the type I receptors ALK4/ALK7 and the type II receptors ActRIIA and ActRIIB to induce Smad2/3 activation (Reissmann et al., 2001; Nadeem et al., 2011). The signalling process is modulated by cripto (TDGF1), which acts as a co-receptor for nodal. The regulation of nodal activity by cripto is complex, as cripto can act both as a membrane-bound and a soluble co-receptor, with opposing effects on nodal-induced signal transduction (Reissmann et al., 2001; Yeo and Whitman, 2001; Pauklin and Vallier, 2015).
Activin/inhibin subfamily
Activins and inhibins, which belong to the TGFβ subfamily, are versatile proteins with a wide range of physiological functions, including gonadal function, hormonal homeostasis, development, reproduction, and tissue homeostasis (Jones et al., 2006b; Tsuchida et al., 2009; Namwanje and Brown, 2016). Both activins and inhibins are composed of β-subunits, with activins forming dimers of two β-subunits and inhibins forming heterodimers of α- and β-subunits (Pryor-Koishi et al., 2007). Heteromeric complexes of type II receptors (ActRIIA and ActRIIB) and type I receptors (mainly ALK4, but also ALK2 and ALK7) are involved in activin signal transduction (Schneider-Kolsky et al., 2002). Upon binding of activin to the respective type II receptor, the type I receptors are activated and initiate the phosphorylation of intracellular Smad2/3. These newly formed complexes, together with Smad4, are translocated to the nucleus and modulate the expression of specific target genes (Moustakas and Heldin, 2009). Inhibins counteract activin signalling by binding one of the type II receptors to the type III receptor, betaglycan. This interaction binds the II receptors in an inactive complex, inhibiting further signal transduction (Namwanje and Brown, 2016).
GDF subfamily
Within the GDF subfamily of TGFβ signalling, a limited number of factors, in particular GDF8 and GDF11 play critical roles in early placental development and function. GDF8 and GDF11, due to their structural similarities (Wu et al., 2022), primarily use the ActRIIA and ActRIIB type II receptors, which are coupled to the ALK4 and ALK5 type I receptors and activate the Smad2/3 signalling pathways. In addition, their signalling is modulated by extracellular antagonists, in particular follistatin-related protein-3 (FSTL3) (Walker et al., 2017). On the other hand, GDF15, also known as macrophage inhibitory cytokine 1 (MIC-1), is a distant member of the family and its mechanism of action seems not to involve TGFβ receptors activation (Mullican et al., 2017; Olsen et al., 2017). The main effects of GDF15 have been related to metabolic regulation through the regulation of PI3K/Akt/ERK kinases (Rochette et al., 2020).
TGFβ signalling in PE
PE is an inflammatory syndrome, and its incidence is on the rise globally. According to the World Health Organization (WHO), the incidence of PE ranges between 2% and 10% of pregnancies worldwide (Gestational Hypertension and Preeclampsia: ACOG Practice Bulletin, Number 222, 2020). It is a major cause of preterm birth and intrauterine growth restriction (Phipps et al., 2019; Raguema et al., 2020). PE is a complex and heterogeneous condition and the underlying pathophysiology is not yet fully understood (Huppertz, 2008b; Than et al., 2018). The syndrome is classified according to the time of diagnosis, before or after 34-week gestation, and subdivided into early-onset (EO) and late-onset (LO) PE (von Dadelszen et al., 2009; Tranquilli et al., 2013; Wójtowicz et al., 2019). While EOPE and LOPE share a number of common, mainly clinical, characteristics, they have different maternal and foetal outcomes, suggesting different underlying etiologies (Tranquilli et al., 2013; Rana et al., 2019; Parada-Niño et al., 2022a). EOPE is often considered as a more severe form, associated with inadequate early placentation, placental dysfunction, reduced placental volume, intrauterine growth restriction and immune maladaptation, and carries a higher risk of future disease for the mother and offspring (Valensise et al., 2008; Li et al., 2014b, 2022). LOPE, on the other hand, is primarily considered a maternal disorder with milder complications, associated with maternal endothelial dysfunction and is generally associated with a more favourable foetal outcome (Valensise et al., 2008). Despite these differences, the placental-derived molecular mechanisms of EO and LOPE remain unknown, and consequently there is a lack of sufficient molecular criteria to distinguish between the PE subtypes (Valensise et al., 2008; Wu et al., 2019; Stepan et al., 2020). One of the key features associated with PE is impaired trophoblast invasion and endothelial dysfunction, both of which contribute to abnormal placental development (Huppertz, 2008b; Than et al., 2018). Studies have shown that villous trophoblasts of PE placentas develop an immature phenotype, both ultrastructurally and biochemically, compared to normal placentas. In particular, EVT cells in the decidua of PE women have a less invasive and more proliferative phenotype than those from uncomplicated pregnancies (Redline and Patterson, 1995).
Placentation, the process of formation and development of the placenta, is crucial for a successful pregnancy. Early in a healthy pregnancy, EVTs invade deep into the endometrial lining of the uterus and remodel the uterine spiral arteries into highly conductive vessels (Pijnenborg et al., 1991; Vanwijk et al., 2000; Brosens et al., 2011). This remodelling ensures an optimal and high flow of oxygenated blood to the uteroplacental bed, which is critical for placental and foetal development (Pijnenborg et al., 1991; Meekins et al., 1994; Brosens et al., 2011; Lyall et al., 2013). In normal placental development, the spiral arteries undergo significant changes during remodelling, including an increase in terminal luminal diameter and loss of elastic and muscular components. These changes extend into the inner third of the myometrium and result in a loss of vascular smooth muscle condensation near the myometrial decidual junction (Vanwijk et al., 2000; Brosens et al., 2011). However, this process is incomplete in PE. Abnormal placentation in PE is characterized by impaired trophoblast invasion, particularly EVT dysfunction, resulting in shallow invasion and a failure to remodel the spiral arteries (Pijnenborg et al., 1991; Lyall et al., 2013; Lin et al., 2020). Terminal dilatation of the spiral arteries is less extensive and smooth muscle removal is inadequate and does not extend beyond the decidua (Brosens et al., 2011). This failed remodelling of the spiral arteries contributes to placental hypoxia, oxidative stress, and endoplasmic reticulum stress, disrupting normal blood flow patterns and reducing nutrient and oxygen delivery to the placenta (Burton et al., 2009b). The resulting ischaemia and hypoxia of the placenta are important factors in the development of PE. The impaired blood flow and inadequate nutrient supply trigger a cascade of events leading to the induction of the possible pathophysiological features that may later develop into the clinical symptoms of PE (Burton et al., 2009a; Brosens et al., 2011). Inadequate placentation, shallow trophoblast invasion, and impaired immune (Mercnik et al., 2022) and endothelial function (Zhou et al., 2019) in PE emphasize its complexity and the need to understand the underlying mechanisms driving its development.
TGFβ signalling has been implicated in the pathophysiology of PE, but its exact role is still not understood. Several TGFβ ligands and cofactors have been suggested as potential biomarkers in the development of PE. Endoglin (Eng) has received much attention, as elevated levels of soluble Eng (sEng) are found in both the serum and placenta of PE patients. These elevated levels correlate with the severity of the syndrome, and can be detected before the onset of clinical manifestations (Venkatesha et al., 2006; Perucci et al., 2014; Leavey et al., 2016). Different mechanisms have been proposed to the increase in soluble Eng expression in PE. For example, the PE placenta is associated with a deficiency of antioxidants such as heme-oxygenase, superoxide dismutase, and catalase. This deficiency leads to in increased oxidative stress, which in turn increases the production of Eng and secretion of its soluble form in the PE placenta (Gregory et al., 2014). Additionally, oxysterols, agonists of liver X receptors have been shown to induce Eng expression in the placenta affected by PE (Henry-Berger et al., 2008; Margioula-Siarkou et al., 2021). While Eng is best known for its interaction with TGFβ1 and TGFβ3 isoforms, it has the ability to interact with other ligands of the TGFβ superfamily, such as BMPs (Barbara et al., 1999).
The literature on the cellular origin, expression, and function of TGFβ family ligands in PE remains controversial and incomplete (Table 2). Contradictory published data on TGFβ isoform levels in combination with a not always well-defined classification of the PE samples used do not allow a selective role of TGFβ in this pathophysiology. Interestingly in placental tissue, Xu et al. (2016) observed a significant upregulation of the Smad2/3 pathway induced by TGFβ in the placenta of EOPE, suggesting a potential role of TGFβ ligands in the pathology of this condition. TGFβ1 is elevated in the plasma and serum of PE patients (Djurovic et al., 1997; Benian et al., 2002; Hennessy et al., 2002; Shaarawy et al., 2016), and has been associated with an increase in diastolic blood pressure (Benian et al., 2002). Consistent with this, higher levels of active TGFβ1 have been observed in both, EO- and LO-PE subtypes of PE, when compared to normotensive controls (Djurovic et al., 1997). However, Hennessy et al. (2002) found lower serum TGFβ1 levels in women with PE, possibly due to the differences in sampling methods. Similar to TGFβ1, TGFβ2 is elevated in the serum of patients with PE (Shaarawy et al., 2016). The expression of GDF8 and its antagonist FSTL3 was significantly increased in the maternal serum of PE subjects compared to controls (Pryor-Koishi et al., 2007). The upregulation of FSTL3 and GDF8 was also confirmed in the PE placenta, suggesting that the tissue itself may be a source of circulating GDF8 and FSTL3 in PE (Guo et al., 2012). Of note, inhibin A and activin A, but not inhibin B, are elevated in women with PE (Yair et al., 2001; Mylonas et al., 2006). As activin A levels are also elevated in PE placentas, it is reasonable to speculate that secreted placental activin A contributes, at least in part, to these differences observed in the maternal circulation (Manuelpillai et al., 2001). Using an optimized method for co-expression network analysis, Tejera et al., identified a number of TGFβ superfamily genes that are upregulated in PE placenta. In addition to ENG, an increased expression of INHBA (encoding inhibin A) and ACVR1 (encoding the BMP receptor ALK2) was quantified (Tejera et al., 2013). ACVR1, as well as ACVR2A (encoding ActRIIA), was already found to be upregulated in PE placenta and maternal circulation (Inkeri Lokki et al., 2017; Li et al., 2022). In particular, ACVR1 mRNA levels in decidua correlate with the severity of PE (Yong et al., 2014). Furthermore, in a study conducted in a Brazilian cohort, ACVR2A was associated with the development of EOPE (Ferreira et al., 2015). Further analysis using single-nucleotide polymorphisms identified a common maternal PE susceptibility locus on chromosome 2q22-23, that affects ACVR2A expression and contributes to the development of PE (Roten et al., 2008). Despite recent developments, it is still crucial to further elucidate the role of TGFβ signalling and dissect the interactions within the different branches of the TGFβ pathway to gain a deeper understanding of their contribution to the pathogenesis of PE, and to unravel their potential as targets or biomarkers for this critical condition. Given the proposed different subtypes of PE, it is highly recommended to study the TGFβ signalling pathways in early- and late-onset PE cohorts separately in future (Valensise et al., 2008; Wu et al., 2019). This focused approach may deepen our understanding of the disease, elucidate specific mechanisms, and reveal potential therapeutic targets specific to each subgroup.
The TGFβ superfamily and placental cells: from early weeks until pregnancy term
Trophoblast cells
Considering the complex role of the placenta in foetal–maternal communication, it may not be surprising that its diverse functions at different stages of normal and abnormal pregnancies involve the coordinated actions of several cell types. Within the first weeks of pregnancy, the human placenta generates epithelial trophoblasts with diverse biological roles including in the attachment of the conceptus to the uterine wall, the establishment of early histotrophic response, and the adaption of the maternal uterine vasculature. Different types of trophoblasts, including stem cells, progenitors, and differentiated subtypes with multiple functions develop (Knöfler et al., 2019). Cytotrophoblasts (CTs), progenitor cells located in the first trimester placental villi, fuse to form the multinucleated syncytium, which plays a key role in nutrient transport and hormone production (Gauster et al., 2022). Thereafter CTs undergo a differentiation process leading to the formation of EVTs through a process similar to that which is well documented in cancer biology, known as the epithelial-to-mesenchymal transition (EMT) (Gonzalez and Medici, 2014; Davies et al., 2016). During the EMT-like differentiation, EVTs partially lose their well-organized epithelial phenotype and transition to a migratory and invasive mesenchymal phenotype. Key features of this differentiation include changes in cell polarity and adhesion (Kokkinos et al., 2010). Essentially, trophoblasts acquire mesenchymal characteristics by exhibiting migratory and invasive capabilities, upregulation of EMT transcription factors such as Snail, increased Wnt signalling, and downregulation of epithelial polarity genes such as Occludin and ZO1 (Knöfler and Pollheimer, 2013). It is important to emphasize that trophoblasts simultaneously maintain certain epithelial features, characterized by the expression of cytokeratin 7 and the absence of mesenchymal vimentin induction (Davies et al., 2016; Haider et al., 2017). These changes are critical for successful embryo implantation in the early weeks of pregnancy (Davies et al., 2016; Haider et al., 2017). These processes involve the migration of EVT cells into the maternal decidua and the remodelling of the maternal spiral arteries to ensure an adequate blood supply to the developing placenta (Haider et al., 2022). Dysregulation of trophoblast proliferation and EVT invasion may contribute to conditions like PE, placenta accreta, and recurrent miscarriage (Incebiyik et al., 2014). Among the signalling molecules involved in regulating trophoblast function, members of the TGFβ superfamily play significant and distinct roles during the first trimester of pregnancy (Fig. 2). Research on the TGFβ superfamily and trophoblast function at pregnancy term is relatively limited, and further investigation is necessary to gain a comprehensive understanding of the pregnancy progression and its potential implications for maternal and foetal health.
Summary of the TGFβ family induced signalling in EVT. During placentation, the coordinated migration and invasion of EVTs is critical for the maintenance of a successful pregnancy. During the first trimester, CTs undergo an EMT-like process and differentiate into EVTs. These EVTs migrate and invade the decidua and maternal spiral arteries in the myometrium. Highly regulated interactions between EVTs and surrounding cells, including NK cells, macrophages (M0), and stromal cells, play an important role in the immunological acceptance and depth of trophoblast invasion. In a normal pregnancy (A), TGFβ signalling regulates the process of trophoblast invasion. Different ligands, such as BMPs, activins, and GDFs, are involved in promoting EVT invasion and migration. For example, BMP2 can directly or indirectly activate different receptors (ALK2, ALK3, or ALK4), subsequently inducing Smad1/5/8 or Smad2/3 activation, depending on the context of signalling and receptor binding. Activin A binds to ALK4 and activates Smad2/3, leading to the activation of metalloproteinase 2 (MMP2). GDF8 and GDF11 also activate Smad2/3 and downstream MMP2. To maintain a balance in trophoblast invasion, the secretion of inhibitors or molecules that attenuate their activity regulates the activity of trophoblast proteases. TGFβ and Nodal subfamilies are involved in the induction of TIMPs, with TGFΒs signalling through ALK5 and Nodals signalling through ALK7. In PE (B), trophoblasts fail to invade the spiral arteries adequately, resulting in reduced blood flow and impaired utero-placental perfusion. The impaired invasion appears to be the first change on cellular level that contributes to the pathogenesis of PE. TGFβ1 inhibits cell invasion through ALK5 and Smad2/3 induction, and contributes to the upregulation of miR142-3p and Snail expression in PE. Non-canonical ERK1/2 signalling may also be involved in inhibiting invasion by inducing Kiss-1 expression. Interestingly, elevated levels of Activin A in PE trigger apoptosis via the Nodal/ALK7 pathway rather than by inducing MMP expression. Elevated levels of Nodal, on the other hand, do not inhibit EVT migration but rather induce apoptosis via the ALK7 pathway. M0, macrophages.
TGFβ signalling in trophoblasts
The balance between factors that promote or inhibit cell invasion is essential for a normal placentation. The TGFβ ligands TGFβ1, TGB2, and TGFβ3, exert several biological effects in trophoblasts and in the regulation of placental invasion (Jones et al., 2006b). As mentioned above, TGFβs bind to the type I receptor ALK5 and activate the Smad2/3 signalling cascade (Budi et al., 2017). The expression of some TGFβ signalling components, Smad2/3/4, TGFβRI, TGFβRII, TGFβ1, and TGFβ2, is highest in trophoblasts during the first trimester and decreases during pregnancy (Xuan et al., 2007). All three forms of TGFβ contribute, at least in part, through different mechanisms to the inhibition of trophoblast invasion and migration (Irving and Lala, 1995; Caniggia et al., 1999; Karmakar and Das, 2002; Tse et al., 2002; Jones et al., 2006b). Consistently, TGFβ3 expression decreases around the ninth week of gestation, correlating with a switch from trophoblast invasion and migration to a proliferative state, characterized by increased fibronectin synthesis (Caniggia et al., 1999). For successful invasion, trophoblasts secrete proteases such as matrix metalloproteases (MMPs), urokinase-type plasminogen activators (uPA), and cysteine proteases such as cathepsins (Lash et al., 2005; Li et al., 2021). The activity of trophoblast proteases is tightly regulated by natural inhibitors (Librach et al., 1994; Karmakar and Das, 2002), such as plasminogen activator inhibitor 1 (PAI-1) and tissue inhibitors of metalloproteinases (TIMPs). TGFβ1, TGFβ2, and TGFβ3 induce the activation of PAI-1 and TIMPs (Lash et al., 2005), thereby increasing cell adhesion to the extracellular matrix (ECM) (Irving and Lala, 1995). Local inflammatory mediators, such as IL1β and TGFβ1, are balanced to further regulate the activity of these enzymes. In this sense, IL-1β promotes the expression of uPA and MMP9, thereby promoting invasion, whereas TGFβ1 upregulates TIMP-1, TIMP-2, and PAI-1, which in contrast inhibit the action of uPA and MMP9 (Laiho et al., 1987; Graham, 1997). Additionally, IL1β induces the expression of MT-MMP1 or MMP14, a collagenase involved in ECM degradation, but exposure to TGFβ1 abolishes the expression of MT-MMP in JEG-3 cells (Librach et al., 1994; Karmakar and Das, 2002).
Another important milestone towards a successful pregnancy is the proper invasion of the EVT. Using RNAseq analysis, Haider et al. (2022) identified that TGFβ/Smad2/3 signalling governs the differentiation of placental EVTs, as demonstrated in primary EVTs and trophoblast organoids. The same group characterized different Smad2/3 phosphorylated residues in trophoblast subtypes. Thus, while C-terminal Smad2/3 phosphorylation was proposed to regulate EVT function and differentiation, Smads2 linker phosphorylation was mainly associated with proliferative primary CTs (Haider et al., 2017). In immortalized HTR8/SVneo cells, cell invasion required the expression of the cell membrane junction protein VE-cadherin. Notably, incubation with TGF-β1 induced the activation of the Smad2/3 signalling and the expression of the Snail and Slug transcription factors, and genetic inhibition of Snail was sufficient to prevent invasion of primary trophoblasts and the HTR8/SVneo cell line (Cheng et al., 2013). Conversely, a recent study showed how TGFβ1 can activate the non-canonical ALK5-ERK1/2 pathway and inhibit invasion in primary EVTs and HTR8/SVneo cells, by inducing downstream kisspeptins (Fang et al., 2022). Interestingly, elevated levels of kisspeptin-1 have been observed in PE placenta, suggesting their potential contribution to disrupted PE pathology (Kapustin et al., 2020). In addition, miR142-3p, which is highly upregulated in PE placenta, has been shown to induce trophoblast invasion by activating the TGFβ1/Smad3 signalling pathway in the HTR8/SVneo cell line (Liu et al., 2019). As mentioned above, in some cell types the TGFβ co-receptor Eng is able to balance Smad signalling between ALK1-Smad1/5/8 and ALK5-Smad2/3. In response to TGFβ1 and/or TGFβ3, Eng was shown to be essential to prevent EVT invasion, and Eng inhibition triggered EVT outgrowth, migration, and fibronectin production in villous explants (Caniggia et al., 1997b). Indeed, TGFβ3 triggers the premature invasive phenotype of the trophoblast, and its downregulation rescues the abnormal invasive phenotype observed in PE placentas (Caniggia et al., 1999; Cowden Dahl et al., 2005). Furthermore, a critical link between TGFβ3 and hypoxia is emerging in normal placental development, where elevated oxygen levels at 10–12 weeks of gestation downregulate HIF-1α and TGFβ3 thereby regulating trophoblast invasiveness (Tal, 2012). However, as demonstrated in JAR, JEG-3, and villous explants in PE, unsuccessful suppression of HIF-1α increases placental TGFβ3 expression, leading to the aberrant trophoblast invasion and persistent placental hypoxia, causing a detrimental loop where chronic/continuous hypoxia further stimulates HIF-1α and TGFβ3 expression (Caniggia et al., 1999; Schäffer et al., 2003; Nishi et al., 2004; Xu et al., 2019).
ECM remodelling is a critical aspect of a successful pregnancy, intricately regulating trophoblast invasion and placental development (Huppertz, 2008b). Furthermore, the composition of ECM determines its effect on trophoblast invasion (Huppertz, 2008a). A study by Xu et al. (2019) investigated lysyl oxidase (LOX) and LOX-like protein 2 (LOX2L) signalling components involved in ECM remodelling and HTR8/SVneo trophoblast invasion in PE placentas. The TGFβ1/Smad3 pathway has been shown to inhibit LOX and LOXL2-dependent induction of collagen production, thereby suppressing the migration and invasion of trophoblasts. Downregulation of LOX and LOXL2 has also been observed in PE placenta (Xu et al., 2019). Additionally, connective tissue growth factor (CTGF), also known as CNN2, is an important downstream target of TGFβ-Smad2/3 signalling and plays a role in extracellular matrix deposition. Elevated levels of CTGF have been detected in the placenta and serum of patients with severe PE and foetal growth restriction, suggesting its potential involvement in regulating of trophoblast invasion in PE. Importantly, genetic knockdown of CTGF partially prevented TGFβ1-induced inhibition of invasion of primary trophoblasts and the HTR8/SVneo cell line (Oh et al., 2009; Chen et al., 2012; Cheng et al., 2017).
In summary, studies have implicated various components of the TGFβ signalling pathway, including Smad2/3, TGFβ receptors, and downstream effectors such as VE-cadherin and kisspeptins, in trophoblast differentiation and invasion. Dysregulation of TGFβ signalling, as seen in PE, may contribute to impaired trophoblast invasion and placental dysfunction.
BMP signalling in trophoblasts
The BMP subfamily is one of the most important signalling pathways in developmental processes, as it is involved in trophoblast differentiation and invasion, cell growth, and apoptosis (Jones et al., 2006b). BMP2 and BMP4 play important roles in placental development and particularly in trophoblast invasion. BMP2 is localized to all trophoblast subtypes and to the decidua during the first trimester of pregnancy, with its levels increasing during this period (Yi et al., 2021). In vitro models such as the use of primary trophoblasts and the cell line HTR8/SVneo have demonstrated the action of BMP2 in regulating human trophoblast invasion and spiral artery remodelling (Zhao et al., 2018b, 2020a,b). During normal pregnancy, there are increased serum levels of BMP2, some of which is secreted by trophoblasts (You et al., 2021). Of note, a recent study identified Hofbauer cells (HBCs) as an additional source of BMP2. Crosstalk between HBCs and trophoblast leads to upregulation of BMP6 via ALK3-Smad2/3-Smad4 signalling in primary trophoblasts, promoting their invasion and vascular mimicry (Deng et al., 2023). In trophoblasts, BMP2 has been shown to regulate EMT-like process, cell adhesion, and invasion (Luo, 2017). To promote trophoblast differentiation, BMP2 regulates the expression of adhesion molecules and genes involved in ECM remodelling in primary trophoblast and HTR8/SVneo (Yi et al., 2021). BMP2 upregulates the expression of ECM-associated genes, including COL6A1, COL7A1, ITGA2, ITGA6, ADAM12, and MMP11 (Yi et al., 2021). The expression of the EMT transcription factor Snail (Luo, 2017) is induced by BMP2 through the induction of lncRNA NR026833.1, which binds to miR502-5p and upregulates Snail expression in primary trophoblasts (You et al., 2021). Snail, in turn, induces MMP2, a key molecule involved in trophoblast invasion at 6–8 weeks of gestation (You et al., 2021). In primary trophoblasts and in the HTR8/SVneo cell line, BMP2 also promotes invasion and endothelial-like tube formation through an ID1–IGFBP3–Slug axis (Zhao et al., 2020b).
The mechanisms by which BMP2 regulates trophoblast invasion are complex. BMP2 binding activates non-canonical Smad2/3 signalling leading to the induction of N-cadherin in primary EVTs and HTR8/SVneo cells (Zhao et al., 2018b). Pharmacological inhibition of ALK2/3, but not ALK4/5/7, effectively diminished the induction of Smad2/3 phosphorylation in response to BMP2 (Zhao et al., 2018a,b). Interestingly, ALK3 is required for BMP2 to induce inhibin A and activin A production in primary EVTs, which may act in an autocrine manner (Zhao et al., 2018a). Other factors, such as the adhesion molecule AMIGO2 and the membrane-bound BMP and activin inhibitor (BAMBI), are involved in BMP2-induced trophoblast invasion and WNT/β-catenin signalling crosstalk in primary trophoblast and HTR8/SVneo (Zhao et al., 2020a; Yi et al., 2021). In EOPE placenta, particularly in trophoblasts, BMP2 levels are decreased, as are the ligands BMP4 and BMP5. In contrast, the expression of the BMP type II receptor BMPR2 is increased (Yi et al., 2021), perhaps as a compensatory mechanism. Consistent with the downregulation of BMP2 mRNA levels in EOPE trophoblasts (Yi et al., 2021), the expression of lncRNA NR026833.1 is downregulated in EOPE (You et al., 2021). The downregulation of BMP2 mRNA and lncRNA NR026833.1 expression in EOPE suggests potential mechanisms contributing to trophoblast dysfunction in PE.
BMP4 is used to stimulate the differentiation of various human pluripotent stem cells (hPSCs) origins, including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), into different trophoblast cell models (Kojima et al., 2017; Jang et al., 2022; Soncin et al., 2022). To introduce BMP4 to hPSCs induces the formation of self-renewing trophoblast stem cells (TSCs), which possess the capability to undergo further differentiation into EVTs and ST (Jang et al., 2022; Soncin et al., 2022). Notably, Kojima et al. (2017) have demonstrated that the concentration-dependent stimulation of BMP4 in human-induced pluripotent stem cells (hiPSCs) leads to the direct differentiation of cells into EVTs and STs without the self-renewing TSC stage. Due to the challenges of obtaining human first trimester tissue samples and the potential biological differences between species, differentiated hPSCs have been used as an alternative model to study trophoblast biology. Successful differentiation of hPSCs into trophoblast-like stem cells requires the inhibition of activin/nodal signalling by BMP4 stimulation (Xu et al., 2002). Furthermore, inhibition of activin/nodal signalling via BMP4 activates FGR signalling, leading to hPSC differentiation into the β-human chorionic gonadotropin (βhCG) hormone-secreting multinucleated ST (Sudheer et al., 2012). In particular, BMP4 activates amniotic and/or mesodermal markers under certain conditions. Therefore, its utilization as a differentiation agent remains controversial, and different models and protocols have been proposed (Roberts et al., 2018; Knöfler et al., 2019; Io et al., 2021).
In summary, the BMP subfamily, particularly BMP2 and BMP4, plays a critical role in placental development, trophoblast invasion and differentiation. The understanding of BMP signalling in PE trophoblasts remains poor, and further studies are needed to clarify the role of BMPs in trophoblast function.
Nodal signalling in trophoblasts
Nodal is a multifunctional cytokine, of key importance during mammalian embryonic development, and involved in the regulation of placental development by inhibiting trophoblast proliferation, EVT formation, and EVT invasion of the decidua (Sarkar et al., 2015). During early pregnancy, nodal and ALK7 are expressed in villous and EVT cells, and their levels are strongly upregulated in PE placentas (Nadeem et al., 2011). In a healthy pregnancy, high levels of nodal/ALK7 inhibit trophoblast proliferation and invasion, and may contribute to syncytialization. Subsequently, reduced levels of nodal/ALK7 allow trophoblast proliferation and invasion. Mechanistically, in HTR8/SVneo and villous explants, Nodal/ALK7 activates the Smad2/3 signalling cascade and upregulates TIMP-1, which subsequently inhibits MMP2 and MMP9 (Nadeem et al., 2011).
Nodal levels are regulated by molecules involved in trophoblast proliferation, such as Lefty (Li et al., 2018), miR-378a-5p (Parada-Niño et al., 2022), miR-376 (Fu et al., 2013), and miR-454 (Adu-Gyamfi et al., 2020). miR-376c inhibits both ALK5 and ALK7 and consequently impairs TGFβ/nodal signalling, leading to increased cell proliferation and invasion. Interestingly, miR376c levels are reduced in placentas and plasma of women with PE (Fu et al., 2013). In PE, where nodal/ALK7 is overexpressed, the activation of Smad2/3 signalling leads to impaired trophoblast invasion and increased apoptosis as shown in HTR8/SVneo and villous explants (Nadeem et al., 2011). The inhibition of JEG-3, JAR, and Bewo proliferation by Nodal is partly mediated by the p27-cyclin E/Cdk2 pathway, resulting in G1-cell cycle arrest (Munir et al., 2004). TGFβ1 can inhibit activin/nodal-induced EVT formation shown in TSC model (Sarkar et al., 2015), while activin A enhances nodal signalling (Yu et al., 2012). Maternal Nodal can induce foeto-placental nodal signalling and modulate the PE susceptibility gene STOX1 through activin A secretion at the maternal interface (Thulluru et al., 2013). As both activin A and nodal have been shown to be elevated in PE, it has been suggested that their interaction promotes trophoblast apoptosis through activation of the nodal/ALK7 signalling in primary trophoblasts and HTR8/SVneo cell line (Yu et al., 2012).
To sum up, nodal plays a critical role in placental development by regulating trophoblast proliferation, invasion, and apoptosis.
Activin and inhibin signalling pathways in trophoblasts
Activins and inhibins are members of the TGFβ subfamily and have a similar structure (Pryor-Koishi et al., 2007). Activin A is the predominant form of activins in pregnancy and is expressed in the syncytiotrophoblast (ST) and underlying CT layers throughout pregnancy. Activin A and inhibin A are secreted by the placenta, decidua, and foetal membranes and are found in the maternal circulation, amniotic fluid, and umbilical cord blood (Florio et al., 2004). Its type I receptors, ALK4 and ALK7, are expressed in the ST during the first and second trimesters and predominantly in the ECs of the villi during the third trimester (Schneider-Kolsky et al., 2002).
Activin A is highly involved in the placentation, acting in an autocrine or paracrine manner, being expressed in the newly formed ST and/or playing a role in the aggregation and syncytialization of CT cells. In the first trimester, activin A stimulates primary trophoblast differentiation towards an invasive phenotype (Bearfield et al., 2005). Villous explant studies have demonstrated that activin A promotes CT outgrowth and fusion, EVT formation, and EVT invasion into the decidua (Caniggia et al., 1997a). To facilitate the foeto-maternal interface, CT differentiate towards the invasive and migratory EVT phenotype in a complex and regulated EMT-like process (Kokkinos et al., 2010). Molecules of the TGFβ superfamily, including activin A, are recognized as potent inducers of the EMT-like process (Li et al., 2014c; Tzavlaki and Moustakas, 2020). Indeed, BMP2 can induce the production, of activin A, leading to the upregulation of N-cadherin, an important marker of EMT (Zhao et al., 2018a,b). Activin A mediated invasion of primary EVTs and HTR8/SVneo is linked to the ALK4-Smad2/3 signalling pathway, leading to the activation of either Snail or Slug and their downstream molecules MMP2, MMP9, and MMP26 (Li et al., 2015a; Adu-Gyamfi et al., 2020; Zhu et al., 2021). Interestingly, excessive levels of activin A can induce apoptosis via the nodal-ALK7 pathway rather than promoting MMP expression (Adu-Gyamfi et al., 2020).
Notably, as shown in primary EVTs and HTR8/SVneo during trophoblast invasion, both TGFβ1 and activin A are capable of signalling through ALK5 and ALK4 receptors, respectively, thereby inducing the Snail transcription factor. Of note, TGFβ has been shown to activate Smad-independent signalling pathways, including MAPK (ERK, p38, and JNK), PI3K, and Rho-like GTPases, whereas activin A preferentially signals via Smad dependent pathways (Matsuzaki, 2011; Li et al., 2015a; Zhu et al., 2021).
Inhibin A, produced by the CT, acts as a feedback mechanism in EVT invasion by antagonizing the production of activin A-activated metalloproteinases (MMP2, MMP7, and MMP9) (Jones et al., 2006a; Adu-Gyamfi et al., 2020). Follistatin, produced by the developing placenta, is an activin A soluble antagonist that regulates EVT outgrowth in villous explants (Caniggia et al., 1997a; Petraglia et al., 1994). Another regulator of activin A is follistatin-related protein (FLRG), which is primarily expressed by the decidua and placental ECs (Ciarmela et al., 2003). FLRG levels are increased in the ST of PE placentas and are associated with low birth weight (Pryor-Koishi et al., 2007). Although follistatin and FLRG give similar signals, their expression differs during pregnancy. While follistatin expression decreases throughout the pregnancy, FLRG expression continues to increase until delivery (Wang et al., 2003; Pryor-Koishi et al., 2007). In PE, placental levels of both activin A and inhibin A are elevated (Yi et al., 2021). Interestingly, hypoxia can enhance the activity of both Inhibin A and Activin A, but their expression is not influenced under low oxygen levels in placental cells (Manuelpillai et al., 2003), suggesting that triggers other than hypoxic conditions may modulate the expression of activin and inhibin ligands during PE.
Both TGFβ1 and activin A are involved in the regulation of βhCG production by primary trophoblasts, although their effects may vary depending on the signalling pathways that they induce and the stage of pregnancy. TGFβ1 has an inhibitory effect, whereas activin A stimulates the expression and secretion of βhCG (Song et al., 1996). The effects of activin A are tightly controlled by the antagonistic effects of inhibin A (Petraglia et al., 1989).
GDF signalling cascade
Among the GDF subfamily of the TGFβ signalling, only a few factors have been identified to play a role in the development and function of the early placenta, including GDF8, GDF11, and GDF15. GDF8, also known as myostatin, and its receptor ACVR2B are localized in EVTs in the first and third trimester placenta and contribute to the regulation of normal placentation, by inducing proliferation and migration of primary EVTs and HTR8/SVneo (Peiris et al., 2014). Interestingly, GDF8 can induce the expression of FSTL3, an antagonist of GDF8 signalling, to further enhance invasion in primary EVTs and the HTR8/SVneo cell line via the ALK5-Smad2/3 pathway (Xie et al., 2020). Additionally, the activated ALK5-Smad2/3 pathway induces the expression of MMP2 in HTR8/SVneo cell line (Fang et al., 2021), thereby stimulating EVTs invasion, providing insight into early placental development, as MMP2 is primarily active between 6–8 weeks of gestation.
GDF11, which is structurally similar to GDF8, induces cell invasion by using the ALK4 and ALK5 type I receptors and subsequently activates Smad2/3 and the expression of MMP2 in primary EVTs and in HTR8/SVneo cells (Wu et al., 2022). In addition, GDF11 regulates the transcriptional regulator ID2, which is required for MMP2 expression and invasion of EVTs (Wu et al., 2022).
Another member of the GDF subfamily, GDF15, also known as MIC-1, is abundantly expressed in the placenta, and has been localized to the CT and ST in particular (Sugulle et al., 2009). Using the HTR-8/Neo cell line and villous explants, fragile X-related protein 1 (FXR1) was been shown to regulate GDF15 expression (Hong et al., 2022). The maturation and processing of GDF15 are mediated by the activity of MMP26, which is co-localized on villous and EVT cells. GDF15 has been shown to promote EVT apoptosis and inhibit proliferation in the HTR8/SVneo cell line (Li et al., 2014a). However, the mechanisms by which GDF15 modulates TGFβ signalling responses in the placenta remain unclear.
In summary, GDFs are mainly involved in the regulation of EVT invasion. Indeed, the exact mechanisms and the significance in healthy and pathological pregnancies are still unknown and require further investigation.
Trophoblast models to study TGFβ signalling
To comprehensively study TGFβ superfamily signalling in trophoblasts, several placental in vitro and ex vivo models are used, each with unique strengths and limitations (Table 3). These models, including primary trophoblasts, cell lines, placental explants, TSCs, organoids, placenta-on-chip, and animal models, provide valuable insights into trophoblast biology (reviewed in Horii et al., 2020; Sheridan et al., 2020; Haider and Beristain, 2023; Li et al., 2023b; Liu et al., 2023). The choice of the appropriate cell model is crucial, depending on the experimental design and specific research questions related to trophoblast development, differentiation, and function (Table 4). Despite the challenges in achieving universally reproducible models, understanding these models is essential for interpreting TGFβ-induced placental disorders. Each model has strengths and limitations that must be carefully considered when interpreting the TGFβ signalling dynamics. Given the ethical constraints of studying first trimester placental tissue, continued progress in placental research is essential. In addition, the development of new 2D and 3D models is needed to better mimic the multicellular environment as in vivo and improve our understanding of TGFβ signalling in trophoblasts.
Primary trophoblasts isolated from the first or third trimester placenta are life-like cells for studying TGFβ signalling in placental development, as they retain their original genetic properties (Li et al., 2023b). However, their limited proliferation and lack of self-renewal make genetic manipulation difficult (Tannetta et al., 2008). To overcome this, researchers often combine them with cell lines such as JEG-3, Bewo, JAR, and HTR8/SVneo. Although originated from choriocarcinoma (JEG-3, Bewo, JAR) or transfected with simian virus 40 large T antigen (HTR8/SVneo), these cell lines are often used to study trophoblast differentiation and invasion, because of their ease of maintenance, stability, and reproducibility by genetic manipulation (Li et al., 2023b; Liu et al., 2023). However, it is important to note that immortalized cell lines may have genetic alterations and phenotypic differences compared to primary trophoblasts (Abou-Kheir et al., 2017; Pastuschek et al., 2021). Their in vitro properties may not fully reflect in vivo signalling, although they are genetically manipulated and reproducible (Msheik et al., 2019; Pastuschek et al., 2021). On the other hand, placental explants, retain the in vivo structure for studying cell-to-cell interactions in healthy and pathological pregnancies, such as PE (Li et al., 2023b). When exposed to flow, placental explants retain tissue integrity, further expanding their potential uses (Kupper et al., 2021). However, explants have limitations in terms of difficulties in gene manipulation, lack of self-renewal, and shorter incubation times in static culture (Li et al., 2023b). In addition, explant cultures maintain cells in their tissue context. This offers the advantage to mimic more closely the in vivo situation, but may require additional work to attribute observed differences and changes to a specific cell type within the tissue.
TSCs are emerging as a valuable 2D in vitro model for studying trophoblast biology. TSCs can be derived from two primary sources: first trimester chorionic villi (CT) and blastocysts (Okae et al., 2018). TSCs are cultured in a specific medium containing activin/nodal/TGFβ pathway inhibitors (e.g. A83-01), Rho-associated protein inhibitors (e.g. Y27632), histone deacetylase inhibitors (e.g. valproic acid), Wnt pathway inducers (e.g. CHIR99021), and epidermal growth factor (EGF) (Okae et al., 2018). This facilitates the long-term expansion of CT-TSCs, which show transcriptional similarities to primary trophoblasts. Forskolin induces TSC fusion into syncytia, and upon removal of Wnt activator, TSCs differentiate into invasive matrix-degrading HLA-G+ EVTs (Okae et al., 2018). Alternatively, TSCs can be derived from hPSCs, including ESCs, iPSCs, or expanded potential stem cells (ePSCs) (Li et al., 2023b). As outlined in the earlier section on BMP signalling in trophoblasts, differentiation induced by BMP4 and activin inhibitors yields TSCs with characteristics similar to primary trophoblasts, although HLA expression patterns may differ from primary trophoblast (Xu et al., 2002; Amita et al., 2013; Koel et al., 2017; Roberts et al., 2018; Horii et al., 2019; Sheridan et al., 2021). Of note, TSCs exhibit normal karyotypes, unlimited proliferation, and versatile differentiation potential, making them adaptable for straightforward genetic manipulation, despite their potential genetic heterogeneity (Li et al., 2023b; Liu et al., 2023).
Development of trophoblast organoids provide an efficient in vitro model to study human placental development (Haider et al., 2018; Turco et al., 2018). Trophoblast organoids derived from primary CT cells in early pregnancy show differentiation into EVTs and STs as well as self-renewal capacity (Haider et al., 2018). These organoids secrete placenta-specific peptides such as hCG and GDF15 and closely resemble first trimester trophoblasts (Turco et al., 2018; Sheridan et al., 2021). Cultured on matrigel with TGFβ and BMP signalling inhibitors (A83-01 and Noggin, respectively), EGF, and enhanced Wnt signalling, the organoids show prolonged expansion potential (Haider et al., 2018). Similar to TSCs, removal of the Wnt inducer leads to re-differentiation of ST organoids into HLA-G+ EVTs (Haider et al., 2018). Inhibition of TGFβ supports early invasive EVT development, whereas promotion of TGFβ signalling is critical for mature decidual EVT expression, highlighting the central role of decidual cell-derived TGFβ in controlling the EVT environment (Haider et al., 2022). Trophoblast organoids present notable advantages, including continuous proliferative capacity and the capability to be cryopreserved and thawed (Sheridan et al., 2020). Despite these benefits, a notable limitation involves the inverted architecture of CT and ST within organoids compared to placental villi (Haider et al., 2018). However, this issue can be addressed by cultivating organoids in suspension culture with gentle agitation (Yang et al., 2023). Furthermore, organoids mirror the expression profile of first-trimester placental tissue, potentially limiting their suitability as a model for third-trimester placenta. However, the complex culturing system they require provides an opportunity to study placental pathologies (Chuva de Sousa Lopes et al., 2020; Karvas et al., 2022).
In addition to 3D organoid models, placenta-on-chip models offer precise control of the microenvironment for real-time trophoblast differentiation and placental barrier studies (Lee et al., 2016). While maintaining a placenta-on-a-chip system can increase the costs, the fluid shear stress environment within the system closely mimics in vivo conditions and provides an alternative to static cultures. This system allows the study of co-cultures with different cell types to investigate cell-to-cell communication and the placental barrier (Park et al., 2022; Li et al., 2023b; Liu et al., 2023). Unlike the aforementioned organoids, in these on-chip models, first trimester and term placental cells can be used alike, making them a versatile tool for different stages of pregnancy. Animal models, including rodents and non-human primates, allow the study of in vivo responses and provide critical insight into the systemic effects of signalling pathways within the tissue environment (Li et al., 2023b). Despite their widespread use, it is important to recognize potential limitations. These can arise from species differences in anatomy and physiology, resulting in structural and functional differences in placental architecture (Carter, 2020). In addition, the challenges of genetic modification, higher costs, and ethical concerns should be considered when using animal models (Makris et al., 2016; Marshall et al., 2018).
Endothelial cells
The human placenta is a unique source of ECs that are specifically adapted to support vascular needs of the developing foetus. Indeed, several distinct subtypes of first trimester placental EC have been identified, highlighting their functional and metabolic heterogeneity (Zhou et al., 2019). Upregulation of specific TGFβ signalling components (i.e. Smad1, Smad4, BMPR2) has demonstrated the importance of TGFβ in determining EC subtypes and an angiogenic functions (Li et al., 2023a). In general, the TGFβ family maintains the endothelium in a quiescent state and regulates vascular development and barrier function (Possomato-Vieira and Khalil, 2016). Dysregulated TGFβ signalling, together with other factors, leads to the endothelial dysfunction, increased vascular tone, and altered vascular permeability observed in PE (Venkatesha et al., 2006). Among the TGFβ family members, TGFβ1 is one of the most studied factors in endothelial biology, being involved in angiogenesis, cell migration, and blood vessel formation and repair, by inducing tube formation and sprouting (Lebrin et al., 2004; Li et al., 2023a). Indeed, neutralization of TGFβ1 leads to impaired endothelium-mediated vasodilation and increased expression of surface adhesion molecules, resulting in increased leukocyte adhesion (Walshe et al., 2009). Under healthy conditions, TGFβ induces the activation of Smad2/3 signalling via ALK5 and TGFΒRII (Goumans et al., 2003). In addition, the membrane-anchored co-receptor Eng is able to fine tune this complex, by enhancing the affinity for ALK1 and promoting a mixed receptor complex containing ALK1/ALK5 (Goumans et al., 2003; Lebrin et al., 2004). This complex leads to the activation of downstream Smad1/5/8 signalling, thereby promoting angiogenesis (Goumans et al., 2002; Lebrin et al., 2004, 2005). By stimulating different Smad pathways, ALK1 and ALK5 have opposite effects on the behaviour of ECs (Goumans et al., 2002). While ALK5 induces PAI-1, a negative regulator of proliferation and migration, and consequently mediates the production of fibronectin and collagen (Lebrin et al., 2005), ALK1 induces the expression of ID-1, which promotes EC migration and proliferation (Goumans et al., 2002; Lebrin et al., 2005). The interplay between ALK5 and ALK1 allows fine regulation of endothelial function in physiological and pathological processes.
The dysregulation of certain members of the TGFβ family in placental EC biology has attracted considerable attention, particularly with the regard to Eng, a highly upregulated TGFβ accessory receptor that may be associated with endothelial dysfunction (Venkatesha et al., 2006; Leaños-Miranda et al., 2019). Although Eng is essential for vascular homeostasis, its elevated levels in certain inflammatory pathological conditions, such as in PE, contribute to impaired angiogenesis and aberrant vascular development (Venkatesha et al., 2006). During inflammation, MMP12 and/or MMP14 cleave the extracellular domain of Eng, which triggers the excessive release of soluble Eng (sEng) in ECs (Tzavlaki and Moustakas, 2020). By interfering with the binding of TGFβ1 to its receptors TGFβRI and TGFβRII, sEng interrupts with downstream signalling events including inhibiting eNOS activation and vasodilation (Venkatesha et al., 2006). Further, pro-angiogenic factors, such as angiopoietin 1 (Ang-1), vascular endothelial growth factor A (VEGF-A), and fibroblast growth factor receptor 2 (FGR-2) prevent the release of sEng through Akt signalling, a key upstream pathway of VEGF-A (Cudmore et al., 2012). However, in PE, there is an increase in sEng levels, accompanied by increased sFlt-1 (a soluble form of the VEGFR1, encoded by the FLT1 gene) and decreased Akt phosphorylation, further exacerbating the condition by antagonizing VEGF-A activity and compromising EC (Venkatesha et al., 2006; Cudmore et al., 2012).
The balance between the endothelial BMP and TGFβ signalling pathways is essential in many cardiovascular diseases (Goumans and ten Dijke, 2018). In addition to regulating TGFβ activity, sEng can promote the interaction of BMP9 and BMP10 with ALK1 in EC (Lawera et al., 2019). Interestingly, BMP9 upregulates ENG mRNA expression and protein expression in EC, where Eng modulates TGFβ1 function, highlighting the role of Eng as a mediator between BMP and TGFβ signalling in EC (Venkatesha et al., 2006). Although sEng was initially thought to be a ligand trap for BMP9 signalling, preventing BMP9 signalling (Castonguay et al., 2011), Lawera et al. (2019) have shown that sEng does not inhibit BMP9 signalling but rather binds BMP9 in a complex and increases the affinity for ALK1, thereby enhancing Smad1/5/8 signalling in ECs. Of note, the sEng/BMP9 complex is highly expressed in PE subjects compared with to normotensive controls. Notably, a negative feedback mechanism has been described whereby sEng stimulates BMP4 expression, which is inhibited by the natural BMP antagonist Noggin. Given that BMP4 contributes to endothelial dysfunction, sEng-BMP4 may be an interesting target for hypertension therapeutics (Gallardo-Vara et al., 2020).
Inflammatory conditions, such as those observed in PE, can trigger signalling pathways and molecular changes that induce endothelial-to-mesenchymal transition (EndoMT) (Derada Troletti et al., 2019). EndoMT is characterized by profound morphological, functional, and molecular changes in the endothelial phenotype (Sánchez-Duffhues et al., 2019). The morphological changes are driven by changes in cell polarity and cytoskeletal rearrangement as ECs differentiate into mesenchymal cells (Ma et al., 2021). During the process of EndoMT, the expression of endothelial markers such as CD31, TIE-1, TIE-2, and VE-cadherin is downregulated, whereas the expression of mesenchymal Vimentin, N-Cadherin, and fibronectin is upregulated (Cho et al., 2018). The phenotypic shift is accompanied by functional changes, such as increased cell migration, and decreased barrier function. It should be noted that EndoMT is a gradual, reversible, and dynamic process, often triggered by paracrine molecules (Ma et al., 2020a). TGFβ signalling is one of the most potent inducers of EndoMT (Ma et al., 2021). Specifically, in human umbilical cord vein ECs (HUVEC), TGFβ1 initiates EndoMT through the phosphorylation of Smad3, leading to the development of the mesenchymal phenotype (Evrard et al., 2016). For example, co-culture of trophoblasts derived from PE placentas and HUVEC of healthy placentas had an effect on the disrupted barrier integrity of the ECs. In addition to observed changes in cell polarity and morphology, the disruption was manifested by the loss of VE-cadherin and occludin, key adhesion molecules (Li et al., 2015b; Wang et al., 2004). Understanding the molecular pathways underlying EndoMT in the context of PE will provide new insights into this pathology that may be exploited for therapeutic gain.
The role of foetal sex linked to effects on the placental endothelium and the development of PE is a subject of increasing research interest. Interestingly, a study conducted by Zhou et al. has demonstrated that female HUVECs are more susceptible to the effects of PE, than male HUVECs. This sexual dimorphism of PE affects the endothelium, with PE dysregulating EC migration, impairing vascularization, and disrupting angiogenesis in female foetal ECs. Conversely, in male foetal ECs, PE upregulates genes associated with cell growth failure, blood pressure, and chronic heart failure (Zhou et al., 2019). Remarkably, a manuscript by Zhou showed that TGFβ1 stimulation resulted in increased cell proliferation only in female PE ECs, whereas normotensive controls showed a decreased response. On the other hand, stimulation with TGFβ1 led to a reduction in cell permeability in normotensive female foetal ECs, and not in PE female ECs (Zhou et al., 2019). The identification of sex-specific PE dysregulated gene networks in the foetus has significant implications for early foetal programming and may be associated with the development of cardiovascular disease in children born from PE pregnancies. As endothelial dysfunction is a hallmark of several cardiovascular diseases, an early identification of molecular networks may have a therapeutic potential to prevent long-term cardiovascular risk in individuals exposed to PE during pregnancy.
Immune cells at the foeto-maternal interface and within the foeto-placental unit
Successful pregnancy requires tolerance between the immune system and the allogeneic foetus (Svensson-Arvelund et al., 2015). Key immune populations at the foeto-maternal interface include regulatory T cells (Tregs), natural killer (NK) cells, and decidual macrophages (dM), while placental macrophages (HBCs) are located in the foetoplacental unit. Despite their location, immune cells coordinate their actions by secreting cytokines to establish immunological tolerance to the foetus, and contribute to vascular remodelling, yet maintain tissue homeostasis (Yagel, 2009; Alijotas-Reig et al., 2014; Reyes et al., 2017; Vondra et al., 2023). TGFβ serves as an important immune regulator during pregnancy (Svensson-Arvelund et al., 2015; Yang et al., 2021). In the first trimester, TGFβ signalling is central to promoting differentiation of Tregs, regulating NK cell function and maintaining the balance between M1 and M2 macrophages at the foeto-maternal interface (Yang et al., 2021). Furthermore, during the third trimester of pregnancy, TGFβ is essential for maintaining tissue homeostasis by regulating an anti-inflammatory phenotype of HBCs (Schliefsteiner et al., 2017, 2020; Mercnik et al., 2022). Perturbances in these regulatory mechanisms are associated with pregnancy complications such as PE or miscarriage (Reyes and Golos, 2018; Yang et al., 2021). Understanding these immune cell interactions and functions is critical to understanding the immune dynamics during pregnancy and their impact on a successful and healthy outcome.
Regulatory T-cells
Tregs are pivotal to a successful pregnancy outcome, as they orchestrate maternal tolerance by ‘education’ of other immune cells (Alijotas-Reig et al., 2014). They exist as thymus-derived and peripherally induced Tregs. In pregnancy, specific recruitment of peripheral maternal Tregs to the decidua seems to take place (Tilburgs et al., 2009); after an initial influx of Tregs to the decidua in the first trimester, where Tregs make up about 10% of the leukocyte population, the Treg density plateaus and even decreases again towards term (Williams et al., 2009).
Specifically for the induction of peripheral Tregs, TGFβ is needed (Wilczynski et al., 2008; Yang et al., 2021). FOXP3 expression is hallmark of Tregs, although FOXP3− subsets exist (Krop et al., 2020). TGFβ1induces the production of CD4+/FOXP3+ Tregs, and these Tregs can in turn secrete TGFβ1and participate in immune regulation by acting on other cells and themselves, in para- and autocrine manners (Huang et al., 2020). Of note, several foeto-placental cell types appear to be able to induce Tregs via TGFβ. Ma et al. (2020b) found that endovascular EVT released TGFβ which was able to induce maternal Treg formation. Oettel et al. (2016) demonstrated that even umbilical ECs release TGFβ which turned maternal T-cells into Tregs, and observed interesting sex-specific differences, with female HUVECs showing a stronger capacity to do so.
Furthermore, TGFβ balances the ratio of Tregs to other T-cell populations; specifically, a tight ratio between Th17 cells and Tregs at the foeto-maternal interface must be maintained for successful pregnancy, as observed from shifts in the Th17/Tregs ratio in patients with recurrent spontaneous miscarriages, and TGFβ is an important factor in its regulation (Wu et al., 2014). A similar shift in TH17/Treg subsets has been observed in PE too, but was not linked to TGFβ in this study (Eghbal-Fard et al., 2019).
In women suffering from PE, changes in Treg frequency and distribution between decidual tissue and periphery have been found (Sasaki et al., 2007). Similar to NK cells, most research on Tregs focusses on Tregs accumulating in the decidua. Quinn et al. (2011) specifically investigated decidua from early-onset as opposed to late-onset PE and controls, and found that Tregs were lowest in EOPE. Little data are available as to the presence Tregs within villous tissue. In a recent histopathological study of different placental pathologies, it was found that FOXP3− reactivity within villous tissue was reduced in PE and IUGR compared to controls, while being increased in gestational diabetes (Emirdar et al., 2021).
In summary, Tregs are pivotal for establishing maternal tolerance during pregnancy, orchestrating the immune balance through recruitment and modulation via TGFβ signalling. Imbalances in Treg populations observed in PE, possibly linked with TGFβ signalling, emphasize the crucial role of Treg regulation in maintaining a successful pregnancy.
NK cells
NK cells are an integral part of the endometrial innate immune milieu and play an important role in maintaining pregnancy (Yagel, 2009). During pregnancy, the majority of leukocytes are decidual natural killer (dNK) cells. These cells are recruited from the periphery in a CXCR4/CXCL12 dependent manner by EVT cells (Hanna et al., 2003) and then are converted to CD56^bright^/CD16− cells by TGFβ, which is secreted, for example, by decidual stromal cells (Keskin et al., 2007). dNK cells exhibit low cytotoxicity compared to other NK cell subsets and rather contribute to vascular remodelling at the foeto-maternal interface by autocrine release of TGFβ, along with VEGF, angiopoietin, and MMP2 and MMP9 (Lash et al., 2006; Fraser et al., 2012). PE is associated with abnormal TGFβ levels in the decidua (Yang et al., 2021); these elevated levels serve as influential factors modulating dNK cell phenotype and function. Abnormal TGFβ levels, particularly TGFβ1 of Tregs, can disrupt dNK cell function and potentially influence the pathophysiology of PE (Zhang et al., 2019). Further comprehensive insights on dNK cells, particularly their involvement in PE, can be found in recent reviews (Liu et al., 2021; Wei and Yang, 2023). Here we have focused on understanding the role of TGFβ signalling in the context of dNK cells.
However, the plethora of research on NK cells in pregnancy has not focussed on NK cells in the decidua or placenta, but on peripheral NK cells in maternal (and less frequently, foetal cord) blood. For the maternal circulation, there are conflicting data on whether the number of peripheral NK cells is increased, decreased, or maintained at PE, and whether these NK cells are more, less, or as cytotoxic as their counterparts from healthy pregnancies (reviewed in Wei and Yang, 2023). Concerning the foetal circulation, two independent studies supported that in cord blood from PE neonates, activated NK cells appear to be expanded compared to regulatory NK cells, thus initiating an inflammatory response (Sohlberg et al., 2014; Loewendorf et al., 2015). However, neither of these studies looked specifically at TGFβ or BMP signalling and are only mentioned here for completeness.
Macrophages
In pregnancy, placental macrophages are a heterogeneous population of immune cells, involved in the maternal–foetal immune regulation, placental cell invasion, angiogenesis, and tissue remodelling (Houser et al., 2011; Reyes and Golos, 2018). Two main types of macrophages are present at all stages of pregnancy. The first type is the decidual macrophages (dM), located in either in the decidua basalis (dBAM) or in the decidua parientalis (dPAM), and the second type are the foeto-placental macrophages, called HBCs (Houser et al., 2011). Regardless of their specific location, their main function is to act as gatekeepers of placental homeostasis (Aneman et al., 2020). Macrophages are keen to polarize into different polarization states in response to microenvironmental stimuli, namely pro-inflammatory M1 and anti-inflammatory M2 (Murray and Wynn, 2011; Murray et al., 2014). The polarization state determines functionality of HBCs, and both dM and HBCs shifts during pregnancy as a function of placental requirements. As the pregnancy progresses, both dM and HBCs develop specific M1/M2 phenotypes, with macrophages in the first trimester tending to exhibit a more pro-inflammatory polarization (Mezouar et al., 2021), but there is a shift towards M2 as pregnancy progresses towards term (Yao et al., 2019). Inappropriate macrophage activation and polarization has been associated with pregnancy disorders such as PE, recurrent pregnancy loss, or chorioamnionitis (Reyes and Golos, 2018). During the first trimester, activated dM secrete a variety of molecules, including tumour necrosis factor (TNF)α and TGFβ1, which may affect regulatory functions of surrounding trophoblasts. In PE, an accumulation of activated dM is observed in proximity of the spiral arteries (Ning et al., 2016). This suggests a potential role for dM in inhibiting trophoblast invasion. M2 polarized dM, secrete high levels of TGFβ1. As discussed above, TGFβ1 is a potent inhibitor of trophoblast invasion and affects the production of MMPs, which are enzymes involved in tissue remodelling (Ning et al., 2016). Conversely, the secretome of EVTs has been shown to regulate the function of dM. In a study by Vondra et al. (2023), the secretome of EVTs containing TGFβ induced the proliferation of dBAM, but not dPAM, through the upregulation of macrophage colony stimulating factor (M-CSF). Additionally, factors secreted by EVT such as TGFβ, M-CSF, and IL10 have been implicated in the regulation of M2 polarization of dM (Aldo et al., 2014; Svensson-Arvelund et al., 2015). Trophoblast-derived IL6 has also been shown to induce a specific M2 polarization of dM by activating the STAT3 signalling, resulting in high expression of CD206, CCL18, IL10, and TGFβ (Ding et al., 2021). Furthermore, abnormal levels of decorin, a molecule produced by first trimester dM, have been associated with the development of pregnancy related disorders such as PE, recurrent pregnancy loss, and foetal growth restriction (Nandi et al., 2016). Decorin regulates the toll like receptor 2 (TLR2)/TLR4-MyD88-NF-κB signalling pathway (Wang et al., 2022), which alters mitochondrial metabolism, and promotes a pro-inflammatory M1 phenotype switch of dM, and reduced TGFβ secretion (Wang et al., 2022).
In summary, studies performed during the first trimester highlight the dynamic interaction between EVTs and dM, where both cell types produce high levels of TGFβ and mutually influence each other’s function. Investigating the interplay between EVTs and dM and their modulation of the TGFβ signalling may provide insights into the early pathogenesis of PE, where TGFβ signalling is impaired.
During the third trimester of pregnancy, TGFβ acts as vital immunoregulator, preserving the homeostatic phenotype of macrophages (Svensson-Arvelund et al., 2015). TGFβs, as major drivers of M2 polarization, are regulators of the function of dM and HBCs (Murray et al., 2014). Both, dM and HBCs constitutively express TGFβ1 (Schliefsteiner et al., 2017, 2020; Pavlov et al., 2020; Mercnik et al., 2022). While dMs only produce the pro-form of TGFβ1, HBCs also secrete the active form of TGFβ1 (Schliefsteiner et al., 2017; Pavlov et al., 2020; Mercnik et al., 2022). In addition, dPAM and dBAM also secrete TGFβ2 (Vondra et al., 2023). HBCs secrete TGFβ2 and TGFβ3, although TGFβ1 is the major ligand secreted by HBCs (Pavlov et al., 2020). The contribution of TGFβ to the tolerogenic and homeostatic functions of HBCs has been demonstrated particularly in the context of infections and inflammatory pathologies that occur during the pregnancy (Azari et al., 2021). For example, during infection with Lysteria monocytogeneses, HBCs undergo pro-inflammatory reprograming. At the same time however, HBCs increase the secretion of anti-inflammatory TGFβ1, which serves to prevent maternal anti-foetal adaptive immunity and further promotes the anti-inflammatory environment (Azari et al., 2021). Additionally, the immunoregulatory actions of TGFβ1 by HBCs have been found to limit HIV-1 replication in HBCs when exposed to exogenous TGFβ (Johnson and Chakraborty, 2012). Furthermore, the increased secretion of TGFβ1 in EO—and LOPE HBCs is associated with the maintenance of anti-inflammatory phenotype of both inflammatory conditions, as TGFβ is widely recognized for its role in resolving inflammation (Mercnik et al., 2022).
These findings highlight the important role of TGFβ signalling in modulating the immune responses and maintaining the immune homeostasis in HBCs during the pregnancy and in providing protection against excessive inflammation such as in PE.
Conclusion and future perspectives
We have summarized how TGFβ signalling plays an essential spatio-temporal role in regulating the function of all placental cells, including trophoblasts, foeto-placental EC, and immune cells. TGFβ ligands contribute to processes such as trophoblast invasion, vascularization, immune tolerance, and tissue remodelling, to ensure successful placental development during pregnancy (Dietrich et al., 2022; Li et al., 2023a). Dysregulation of TGFβ signalling has been linked to the pathogenesis of PE, a complex disorder characterized by shallow trophoblast invasion, defective vascular remodelling, decreased uteroplacental perfusion (Haider et al., 2017), and impaired EC (Zhou et al., 2019) and macrophages (Mercnik et al., 2022). Despite significant progress, there are still many unknowns and challenges in understanding TGFβ signalling in the context of PE (Fig. 3). In trophoblasts, the specific mechanisms by which TGFβ ligands regulate invasion and EMT-like process remain poorly understood. In EC, the precise role of TGFβ signalling in impaired vascular remodelling, angiogenesis, and possible endothelial-to-mesenchymal transition (EndoMT) requires further investigation. TGFβ serves as an important immune regulator during pregnancy and plays a central role in Treg differentiation and regulation of NK cell function, which is poorly understood. In macrophages, the influence of TGFβ signalling on polarization, cytokine production, and tissue remodelling during PE remains to be elucidated. Further research is needed to unravel the dysregulation of TGFβ ligands, the regulation of receptor activation, the crosstalk with other signalling pathways, and the epigenetic regulation in PE. Of note, distinguishing between the different clinically manifest subtypes of PE is critical to advancing our understanding of TGFβ signalling in this syndrome. Understanding the interactions and dysregulation of TGFβ signalling in these cell types is essential to uncover the underlying mechanisms that contribute to the development of PE. This knowledge may facilitate the development of improved in vitro and in vivo models to study PE and identify potential therapeutic targets within the TGFβ signalling pathway.
The unequal importance of TGFβ signalling in PE-compromised placental cells. The figure provides a cross-sectional representation of the placental villous structure, illustrating a comparison between a healthy (CTR) placenta and a preeclamptic (PE) placenta. The main placental cell types, including trophoblasts, ECs, and immune cells, are depicted within the structure, highlighting their specific known roles in the respective placenta. Question marks (?) indicate the unknown and challenging aspects of TGFβ signalling in these specific cell types. These questions relate to the precise mechanisms and effects of TGFβ signalling in trophoblast function, the impaired vascular remodelling and angiogenesis of ECs, and the phenotypic changes observed in immune cells during PE. FB, fibroblasts; HBC M1, M1 polarized Hofbauer cells; HBC M2, M2 polarized Hofbauer cells; ERY, erythrocytes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abdollah S , Macías-Silva M, Tsukazaki T, Hayashi H, Attisano L, Wrana JL. TβRI phosphorylation of Smad 2 on Ser 465 and Ser 467 is required for Smad 2-Smad 4 complex formation and signaling. J Biol Chem 1997;272:27678–27685.9346908 10.1074/jbc.272.44.27678 · doi ↗ · pubmed ↗
- 2Abou-Kheir W , Barrak J, Hadadeh O, Daoud G. HTR-8/S Vneo cell line contains a mixed population of cells. Placenta 2017;50:1–7.28161053 10.1016/j.placenta.2016.12.007 · doi ↗ · pubmed ↗
- 3Adu-Gyamfi EA , Ding YB, Wang YX. Regulation of placentation by the transforming growth factor beta superfamily. Biol Reprod 2020;102:18–26.31566220 10.1093/biolre/ioz 186 · doi ↗ · pubmed ↗
- 4Aldo PB , Racicot K, Craviero V, Guller S, Romero R, Mor G. Trophoblast induces monocyte differentiation into CD 14+/CD 16+ macrophages. Am J Reprod Immunol 2014;72:270–284.24995492 10.1111/aji.12288 PMC 4230492 · doi ↗ · pubmed ↗
- 5Alijotas-Reig J , Llurba E, Gris JM. Potentiating maternal immune tolerance in pregnancy: a new challenging role for regulatory T cells. Placenta 2014;35:241–248.24581729 10.1016/j.placenta.2014.02.004 · doi ↗ · pubmed ↗
- 6Amita M , Adachi K, Alexenko AP, Sinha S, Schust DJ, Schulz LC, Roberts RM, Ezashi T. Complete and unidirectional conversion of human embryonic stem cells to trophoblast by BMP 4. Proc Natl Acad Sci USA 2013;110:E 1212–E 1221.23493551 10.1073/pnas.1303094110 PMC 3612666 · doi ↗ · pubmed ↗
- 7Aneman I , Pienaar D, Suvakov S, Simic TP, Garovic VD, Mc Clements L. Mechanisms of key innate immune cells in early- and late-onset preeclampsia. Front Immunol 2020;11:1864.33013837 10.3389/fimmu.2020.01864 PMC 7462000 · doi ↗ · pubmed ↗
- 8Arutyunyan A , Roberts K, TrouléK, Wong FCK, Sheridan MA, Kats I, Garcia-Alonso L, Velten B, Hoo R, Ruiz-Morales ER et al Spatial multiomics map of trophoblast development in early pregnancy. Nature 2023;616:143–151.36991123 10.1038/s 41586-023-05869-0PMC 10076224 · doi ↗ · pubmed ↗