The JMU-SalVac-System: A Novel, Versatile Approach to Oral Live Vaccine Development
Andreas Iwanowitsch, Joachim Diessner, Birgit Bergmann, Thomas Rudel

TL;DR
Researchers developed a new oral vaccine system using a modified Salmonella strain to safely and effectively deliver antigens for mucosal immunization.
Contribution
A novel balanced-lethal system (BLS) ensures plasmid stability in an oral Ty21a-based vaccine platform for human use.
Findings
The JMU-SalVac-system uses a ΔtyrS/tyrS+-based BLS to stabilize antigen delivery plasmids in Ty21a.
Cytosolic and secreted model antigens (mRFP and CTB) were successfully expressed in vaccine strains.
Promoters in pSalVac-plasmids were induced in primary macrophages, demonstrating in vivo functionality.
Abstract
Salmonella enterica Serovar Typhi Ty21a (Ty21a) is the only licensed oral vaccine against typhoid fever. Due to its excellent safety profile, it has been used as a promising vector strain for the expression of heterologous antigens for mucosal immunization. As the efficacy of any bacterial live vector vaccine correlates with its ability to express and present sufficient antigen, the genes for antigen expression are traditionally located on plasmids with antibiotic resistance genes for stabilization. However, for use in humans, antibiotic selection of plasmids is not applicable, leading to segregational loss of the antigen-producing plasmid. Therefore, we developed an oral Ty21a-based vaccine platform technology, the JMU-SalVac-system (Julius-Maximilians-Universität Würzburg) in which the antigen delivery plasmids (pSalVac-plasmid-series) are stabilized by a ΔtyrS/tyrS+-based…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
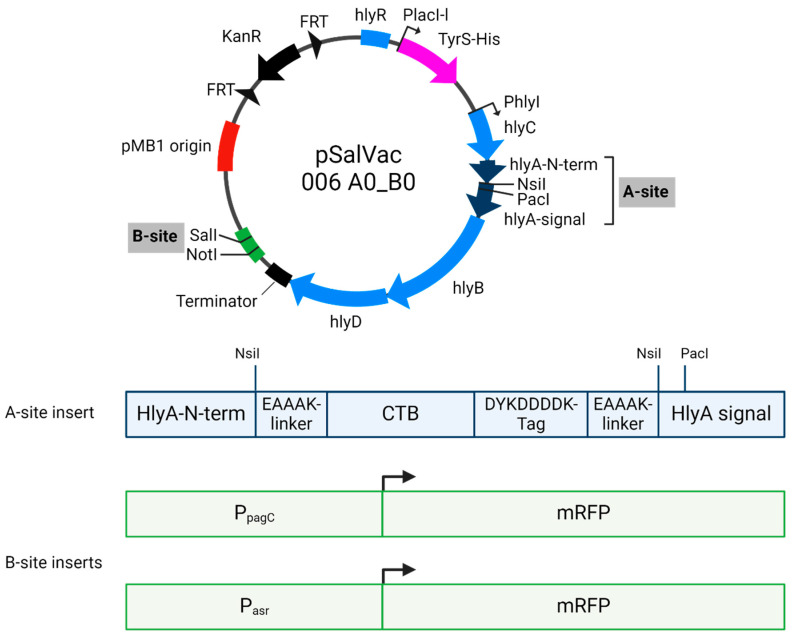 Figure 1
Figure 1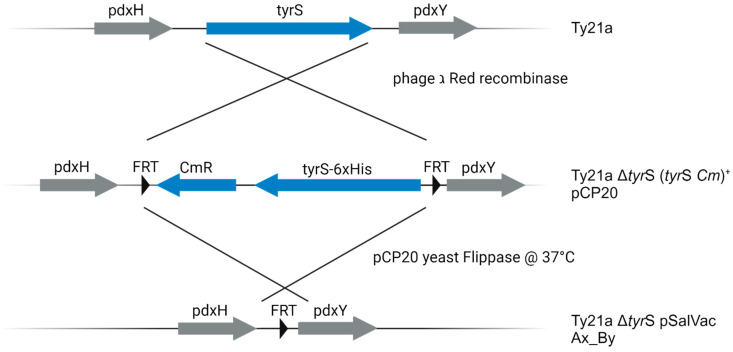 Figure 2
Figure 2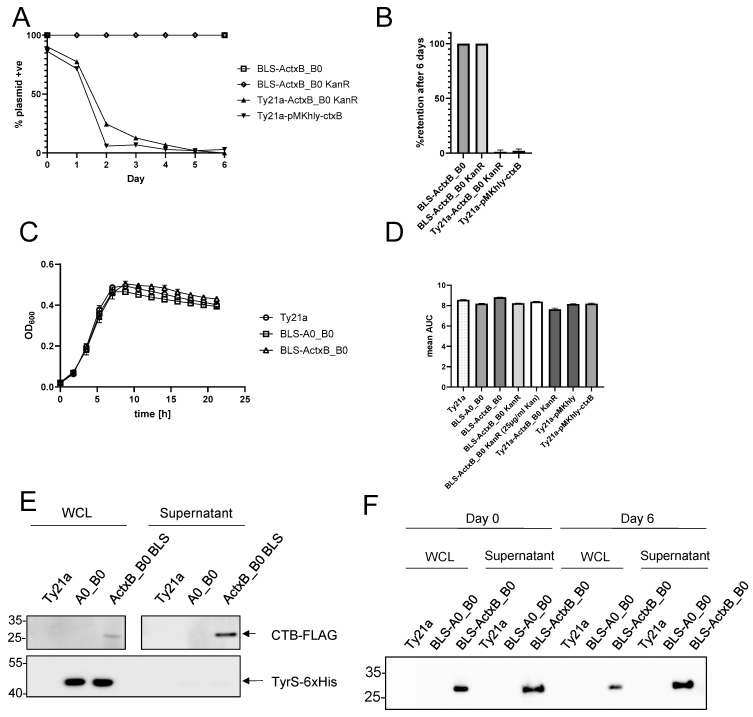 Figure 3
Figure 3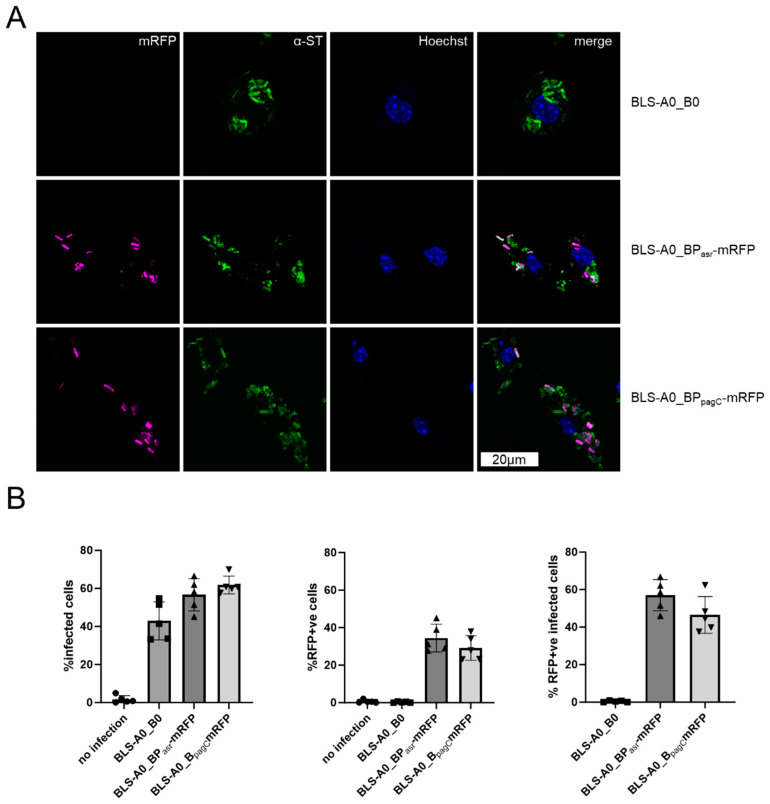 Figure 4
Figure 4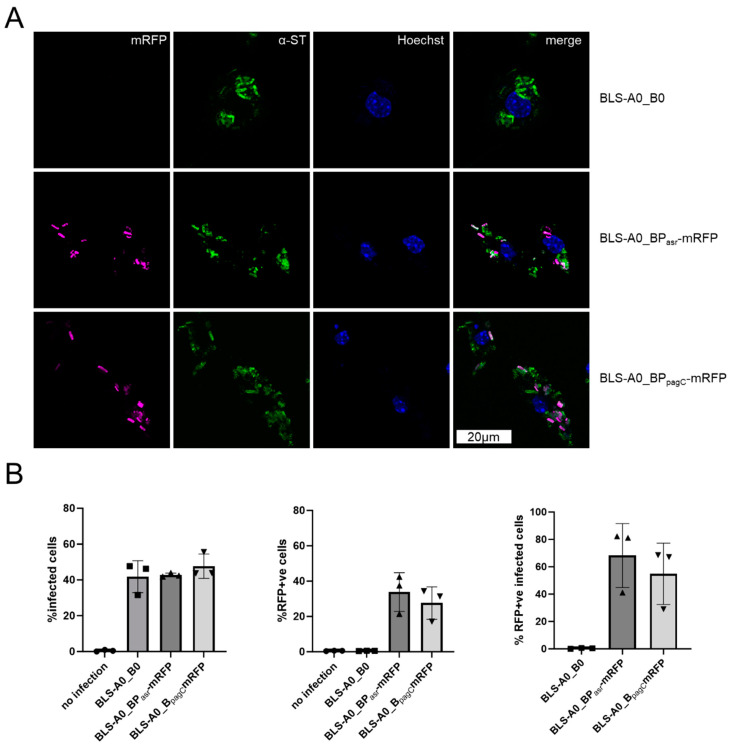 Figure 5
Figure 5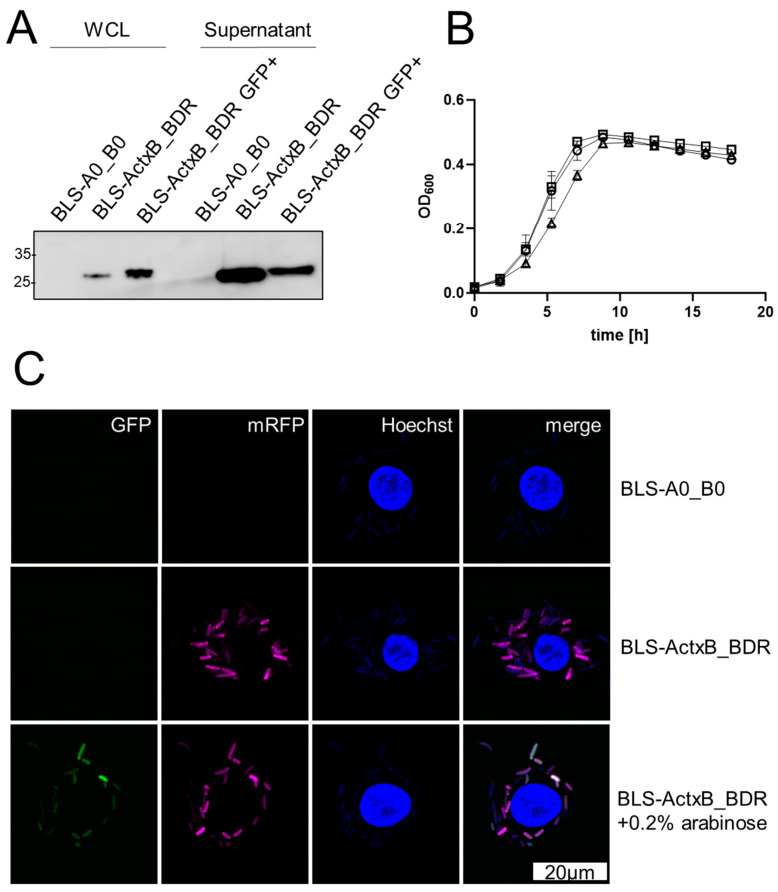 Figure 6
Figure 6- —German Ministry of Science and Education
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRNA Research and Splicing · RNA and protein synthesis mechanisms · Animal Genetics and Reproduction
1. Introduction
An efficient and cost-effective approach to combat mucosal infections is to stimulate the mucosal immune system via live attenuated bacterial vaccines administrated via mucosal routes. One of the key issues in developing a bacterial vaccine system is to find an optimal balance between attenuation of the vaccine strain to ensure safety and sufficiently high expression of vaccine antigens to trigger an immune response. Excessive attenuation often leads to reduced immunogenicity due to reduced fitness and an impaired ability to colonize and persist in lymphoid tissues (reviewed in [1]). In addition, high antigen expression can also have a negative effect on immunogenicity by impairing metabolism and reducing fitness [2]. To develop an effective, safe and stable antigen expression system using live Salmonella bacteria as a vehicle, an optimal ratio between antigen expression and fitness of the vaccine strain must therefore be achieved.
The expression of heterologous antigens in live bacterial vector strains by the incorporation of stable plasmids is a flexible and easy to realize method for the production of new vaccines, especially in comparison to the genomic integration of expression cassettes. The expression of antigens via high-copy number plasmids may allow a higher production of antigens than expression via the genome but may have the disadvantage that it affects the fitness of the vehicle strain. There are different ways to circumvent negative effects of high antigen expression on the fitness of the vaccine strain, e.g., the use of plasmids with lower copy number or the use of promoters that can be induced in vivo (regulated delayed protein synthesis). In addition, suitable sub-cellular targeting can optimize the production of the antigen and the fitness of the vaccine strain and significantly improve the strength and type of immune response (reviewed in [3,4]).
It has been shown that secretion of the vaccine antigens, in contrast to cytosolic expression, increases the immunogenicity of the antigens and additionally reduces metabolic burden on live oral vaccines [4,5]. The hlyA secretion system of Escherichia coli has already been used successfully for secretion of antigens [6,7]. The hly operon consists of four essential components, the transcription enhancer sequence hlyR, the HlyA acylating enzyme HlyC and the inner membrane complex HlyB-HlyD [8]. HlyB transports antigens which have been fused to the hlyA-signal across the inner membrane, while HlyD interacts with TolC in the outer membrane for secretion into the lumen [8]. Actively exported antigens become accessible to the host immune system without the bacteria being degraded [8,9,10].
Antigenicity of the heterologous protein is another key factor for successful vaccination with live vaccines. Uptake and presentation by antigen-presenting cells (APCs) is a decisive step, which may be enhanced by peptide fusions targeting APCs, such as the non-toxic subunit of the cholera toxin CtxB [11]. CTB pentamers bind to monosialotetrahexosylganglioside (GM1) present on gut epithelial cells and APCs, even when antigens are fused to it [12]. Immunization strategies in which CTB is administered as adjuvant either as CTB antigen fusion or as recombinant CTB show enhanced immune responses [13,14]. CTB adjuvants have been classified as safe by the regulatory authorities, as demonstrated by their use in one of the three approved vaccine formulations against Vibrio cholerae, Dukoral^®^.
When using plasmids for antigen expression in vaccine strains, one challenge is to ensure vector stability. Although antibiotics are useful and effective in providing plasmid stability under selective conditions in vitro, their use in vivo is usually not applicable and moreover raises safety concerns in cases where the goal is to generate a product for administration to humans. Even the use of antibiotics in fermentation cultures bears the risk that the final product is contaminated with residual antibiotics that in turn could potentially cause an allergic reaction in sensitive individuals [15]. Furthermore, regulatory agencies recommend avoiding antibiotic resistance genes in plasmids due to the risk of antibiotic resistance being transferred to other organisms in the environment, especially pathogens [16,17].
The stabilization of plasmids in live vaccines must therefore be achieved by other means, in particular by balanced-lethal host-vector systems (BLS), where, for example, a gene coding for an essential protein is deleted from the host chromosome and complemented on the plasmid. One of the first and most prominent BLSs is the well-studied asd BLS. The protein encoded by the aspartate-β-semialdehyde dehydrogenase (asd) gene is an enzyme involved in the biosynthesis of diaminopimelic acid (DAP) from aspartate. Deletion of asd (Dasd) makes the bacteria auxotrophic for DAP resulting in lysis when the bacteria grow in absence of DAP. By integrating a functionally active asd expression cassette into the plasmid, the chromosomal deletion can be complemented, and the plasmid can be stably maintained in the carrier in the absence of DAP. Since DAP hardly occurs in animal or human organism, the loss of the plasmid leads to lysis of the Dasd bacteria, as they can no longer synthesize peptidoglycan [18]. Although promising results were obtained in a mouse models [19,20,21], clinical trials with a S. Typhi Ty2-derived ∆asd vaccine strains were disappointing [22,23]. It was speculated by Galen et al. that the catalytic activity of Asd could be rescued by a single or few copies of asd, likely leading to suboptimal maintenance of antigen-expressing plasmids [24]. To circumvent this fundamental problem, the authors developed an alternative BLS based on the non-catalytic single-stranded DNA-binding protein (SSB), whose crucial function is to prevent unstable ssDNA intermediates [24]
In addition to proteins essential for the biosynthesis of peptidoglycan like Asd, the thymidylate synthase ThyA, an enzyme involved in synthesis of nucleic acid building blocks was employed for the design of an auxotrophy-based BLS [25].
Based on the considerations above, we developed an antigen expression platform technology (JMU-SalVac-system) in which the antigen delivery plasmids (pSalVac-plasmid-series) are stabilized by a ΔtyrS/tyrS^+^-based balanced-lethal system in Ty21a. The system consists of the chromosomal knockout of the essential gene tyrS, which encodes for the tyrosyl-tRNA-synthetase, and the in trans complementation of this gene on pSalVac-plasmids. The pSalVac-plasmids enable the simultaneous expression of two heterologous proteins. In this study, we demonstrate the stability of the plasmid, the expression of mRFP and the secretion of the mucosal adjuvant CTB from the vaccine strain Ty21a. The model antigens mRFP and CTB are expressed in infected human primary macrophages under the control of in vivo inducible promoters, demonstrating the application of the JMU-SalVac-system in these central immune cells. In summary, we have developed a system in which antigens can be stably, safely and efficiently expressed, indicating that bacterial strains developed with our system have the potential to be used as safe and stable vaccine strains against mucosal infections suitable for use in humans.
2. Methods and Materials
2.1. Rational Design
To ensure 100% maintenance of Ty21a cells with the desired antigen delivery plasmids, we decided to develop an antibiotic resistance gene-free balanced-lethal system that is not based on auxotrophy, but on complementing the chromosomal deletion of an essential gene on pSalVac plasmids. As an essential gene we chose tySfor several specific reasons. The tyrSgene is a single copy gene in the Salmonella genome with a short coding sequence of 1275 bp. Although aminoacyl-tRNA synthetases are evolutionarily very old and highly conserved, some members, such as TyrS, exhibit species-specific recognition. Crucially, prokaryotic and human cytoplasmic tyrosyl-tRNA synthetases cannot acylate their respective counterpart’s tRNA [26], which excludes the possibility of cross-complementation and ensures the dependence of the bacterial carrier on the BLS plasmid. We decided to choose the tyr**S gene from E. coli K-12 instead of the native Salmonella tyrSgene as a recombinant complementing gene on the extrachromosomal vector to reduce the potential risk of unwanted integration during the chromosomal recombination step. The tyrSgene in E. coli is located within the same flanking genes as its counterpart in the Salmonella genome and shows 88% nucleotide identities with the respective Salmonella gene, whereas both gene products have 96% amino acid identity. Furthermore, the E. coli tyrSgene locus has been well studied with regard to promoter structure, regulation of expression and termination of the tyrSgene [27,28]. Preliminary studies of tyrS-6xHis-expression in E. coli under control of its own wildtype promoter showed high levels of synthesis of the TyrS-6xHis protein and a reduction in growth rate (see Supplementary Figure S6). To avoid toxic effects of tyrS-overexpression, we decided to reduce tyrS-expression by placing the gene under the control of a putative weaker promoter. It is known that in E. coli, the LacI repressor which regulates expression of the lactose metabolic genes by binding to the lacO operator sequence [29] is constitutively synthesized at a very low level of about 5 to 10 copies per cell [30,31]. Thus, to reduce the expression on each single plasmid and therefore favor the regulation of expression towards a higher plasmid copy number, the tyrS-6xHis-coding sequence was cloned under the control of a lacI-derived promoter (PlacI-like) and integrated into pSalVac-plasmids.
2.2. Standard Microbial and Molecular Methods Used for Generation of JMU-SalVac Strains
If not noted otherwise, E. coli DH5α (Invitrogen, Darmstadt, Germany) was utilized for subcloning, plasmid amplification and maintenance. S. enterica serovar Typhi strain Ty21a and its ΔtyrS derivatives were used as the basis for the generation of antigen expression strains. Unless otherwise stated, bacterial strains were grown aerobically in LB broth Lennox vegetal (Carl Roth, Karlsruhe, Germany) at 37 °C with rigorous shaking (180–200 rpm), or on LB-Agar (Lennox) vegetal (Roth). Antibiotic selection was performed as required with ampicillin (Sigma-Aldrich), kanamycin (Sigma-Aldrich, Darmstadt, Germany) and chloramphenicol (Sigma-Aldrich) at final concentrations of 100, 25 and 20 µg/mL, respectively. All bacterial strains and plasmids used in this study are listed in Table 1. Primers are listed in Supplementary Table S1. Standard molecular methods were performed following published protocols [32]. PCR-products and digests were purified either with QIAquick PCR Purification Kit (Qiagen) or the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany) following the manufacturer’s recommendations. Plasmids were purified with QIAprep Spin Miniprep Kit (Qiagen) and QIAGEN Plasmid Midi Kit (Qiagen) following the manufacturer’s instructions.
2.3. Construction of the BLS-Recipient Strain S. enterica Typhi Ty21a ∆tyrS(tyrS CmR)+
For the construction of the chromosomal tyrS-knockout we modified the method of “one-step inactivation of chromosomal genes using PCR products” which was described by Datsenko and Wanner, 2000 [33]. As tyrS is an essential gene, a knockout without genetic compensation would be lethal. Therefore, we first inserted a functional TyrS-expression cassette into the PCR-template-plasmid pKD3. For excision, the tyrS expression cassette and the chloramphenicol resistance gene are flanked by two FRT sites. Thus, during recombination, the chromosomal tyrS is not only replaced by a fragment encoding for the antibiotic resistance, but also by a gene encoding E. coli tyrS. In brief, we introduced the tyrS gene upstream of CmR and amplified the resulting FRT-tyrS-CmR-FRT-region with primers featuring homologous overhangs to the genomic tyrS. After electroporation of the PCR product into S. enterica Typhi Ty21a harboring the helper plasmid pKD46, a temperature-sensitive Red helper plasmid which carries the Red recombination system with the phage ג Red recombinase under control of an arabinose-inducible promoter [35], one Cm-resistant transformant was selected and correct integration of the FRT-*tyrS-*CmR-FRT-Fragment replacing the tyrS-locus was confirmed via PCR and sequencing. After removal of the pKD46 plasmid by incubation at 37 °C, the selected clone was made electrocompetent and transformed with the helper plasmid pCP20. The resulting strain (S. enterica Typhi Ty21a ∆tyrS (tyrS CmR)^+^ pCP20) represents the balanced-lethal-system-recipient strain of our JMU-SalVac-system: TyrS complementing pSalVac-plasmids which are intended to be BLS-stabilized are transformed into Ty21a ∆tyrS (tyrS CmR)^+^ pCP20for generation of the final antibiotic resistance gene free JMU-SalVac antigen delivery strains.
2.4. Design and Construction of the pSalVac Antigen Delivery Plasmids
The basic pSalVac antigen delivery plasmid pSalVac 006 A0_B0 KanR (see Figure 1) forms the basis of the various antigen delivery plasmids of the pSalVac Ax_By KanR-series (x and y represent placeholders for different antigen-expressing cassettes) used in this study. It is derived from the first generation pSalVac 001 A0_B0 KanR (Patent No. WO 2022/034221 A1) plasmid which has a pMB1 origin of replication (about 20 copies per cell [39]). For selection in vitro, it has a kanamycin resistance expression cassette (KanR) that is flanked by two sites of flippase recognition targets (FRT-sites [10]). This allows the excision by the site-specific enzyme FLP recombinase, which acts on the direct repeats of the FRT-sites [33]. Functional features of the pSalVac Ax_By KanR- series are two independent expression cassettes for the different adjuvant and/or antigen-fusion proteins. The first expression cassette, named A-site, consists of the transcription enhancer sequence hlyR, the structural genes hlyC, hlyB and hlyD and two short residual sequences of the hlyA gene separated by a single NsiI-restriction site (integration site of in silico designed coding sequence of planned fusion protein). A terminator sequence was introduced after the hlyD coding sequence using complementary primers. As the integration site for the complementing tyrS expression cassette on the antigen delivery plasmids, we selected a region within the hly operon between the enhancing sequence hlyR and hlyC (Figure 1). This region contains an IS2 sequence which has been shown to have no effect on HlyA secretion [40]. The activity of the hlyR enhancer is dependent on the correct spacing from the hlyC orf, however, which is why we chose to replace IS2 with the TyrS cassette. This ensures the correct spacing between hlyR and hlyC, while eliminating the unnecessary IS2 transposon element. A multiple cloning site for the second expression cassette (B-site), was integrated into the unique SalI site of pSalVac 001 A0_B0 KanR. For the generation of the different plasmids of the pSalVac Ax_By series, the NsiI-fragments (x) were cloned into the A-(NsiI)-expression site, whereas the B-site-fragments (y) were cloned into the B-(MCS)-expression site of the pSalVac 006 A0_B0 KanR vector. For this study, we used the inactive cholera enterotoxin B-subunit CTB as model antigen. Rigid EAAAK-linker were used to connect the CTB and the adjacent sequence to facilitate domain formation and improve adjuvant efficacy [41]. Java Codon Adaptation Tool (JCAT) (http://www.jcat.de/ accessed on 24 June 2020) [42] was used for codon optimization of the coding sequences to S. enterica Typhi (strain ATCC 700931/Ty2). The optimized NsiI-Linker-ctxB-Linker-FlagTag-Linker-NsiI sequence was synthesized (Thermo Fisher, Darmstadt, Germany) and introduced into the pSalVac 006 A0_B0 KanR plasmid via In-Fusion cloning (Takara Biotech, Saint-Germain-en-Laye, France) resulting in pSalVac A_ctxB__B0 KanR. For an additional control strain, one FRT site flanking the KanR was deleted from this plasmid via In-Fusion to prevent KanR excision, resulting in the BLS-ActxB_B0 KanR variant after stabilization. For intracellular expression analysis, previously described in vivo inducible promoters P_asr_ (acid induced [43]) and P_pagC_ (low Mg^2+^ [44]) were cloned upstream of mRFP using In-Fusion cloning (Takara Biotech) according to the manufacturer’s protocol (see Figure 1). Promoter sequences were amplified from the Ty21a genome and ribosome binding sides were optimized with according primers using the UTR designer tool (https://sbi.postech.ac.kr/utr_designer/reverse/ accessed on 9 November 2022 [45]). The dual reporter system was amplified from pFCcGi [34] and cloned into pSalVac 006 A0_B0 KanR using In-Fusion cloning. Sequences of all plasmids used in this study were routinely verified by whole-plasmid sequencing (Microsynth, Balgach, Switzerland).
2.5. Generation of BLS-Stabilized Vaccine Strains
The presence of FRT sites flanking the chromosomally integrated (tyrS CmR)^+^ cassette and the kanamycin resistance gene on the tyrS-complementing pSalVac plasmids enables the simultaneous deletion of those loci by flippase, yielding a BLS-stabilized strain which has no residual antibiotic resistance genes (see Figure 2). In contrast to the method described by Datsenko and Wanner [33], not only the FRT-flanking fragment in the chromosome but also the FRT-flanking kanamycin resistance gene cassette in the plasmid had to be eliminated. To assure elimination of all FRT flanked sequences, we established a modified protocol for the elimination step. In brief, electrocompetent cells of BLS-R were transformed with one of the pSalVac antigen delivery plasmids. After 1 h incubation at 30 °C in LB broth without antibiotics, kanamycin/ampicillin/chloramphenicol triple resistant transformants were selected at 30 °C on LB agar plates containing 25 µg/mL kanamycin and 100 µg/mL ampicillin as described [33]. Ampicillin is required for selection of pCP20. KanR, AmpR, and CmR-positive clones were cultivated in LB-Medium containing 100 µg/mL ampicillin and 25 µg/mL kanamycin at 30 °C overnight. The next day, the culture was diluted 1:1000 in LB-Medium containing only 100 µg/mL ampicillin and cultivated for 1 h at 37 °C and subsequently for 1 h at 30 °C. These two steps were repeated at least four times. Incubation at 37 °C leads to the expression of yeast flippase from the pCP20 plasmid [35] to simultaneously eliminate the kanamycin-(plasmid) and tyrS CmR (genome) cassettes, while recovery at 30 °C serves to replenish copies of pCP20, which does not replicate at 37 °C. This approach is an adaptation of the original method of Datsenko and Wanner (2000) [33] which is necessary to quantitatively remove all FRT-flanked sequences (from the chromosome and the plasmid). After the temperature shifts, the cultures were selected on LB-Amp at 30 °C [33] and single colonies were screened for kanamycin sensitivity. Kanamycin-sensitive clones were then inoculated into LB-Medium without antibiotics and shaken at 37 °C overnight to eliminate pCP20. Single colonies were tested for sensitivity to ampicillin, kanamycin, and chloramphenicol. Additionally, the correct excision of the genomic tyrS locus as well as the kanamycin locus on the plasmid were confirmed by colony PCR.
2.6. Plasmid Stability Assay
Plasmid maintenance in vitro was determined by serial passage of bacteria cultures without any selective pressure. In brief, overnight cultures of tested strains were diluted 1:40 in LB-Medium without (only BLS-ActxB_B0) or supplemented with 25 µg/mL kanamycin until mid-logarithmic phase to generate generation 0. Subsequently, these starter cultures were diluted 1:1000 into fresh LB-Medium and cultured to stationary phase. In the same way, bacterial cultures were passaged up to 6 times. Each day, serial dilutions of the strains harboring plasmids with kanamycin resistance gene were plated on TS-agar plates without antibiotic selection and incubated at 37 °C for 18–24 h to obtain single colonies. At least 100 colonies per day and strain-harboring plasmids with kanamycin resistance gene were selected randomly and grown on fresh TS agar plates containing 25 µg/mL kanamycin and on TS agar without antibiotics. In case of the investigated antibiotic resistance-free BLS-stabilized vaccine strain (BLS-ActxB_B0), cultures of day 0 and 6 were serially diluted and plated on TS agar plates. After incubation overnight at 37 °C, at least 100 colonies were picked on TS agar. The presence of the BLS-stabilized plasmid (ΔKanR) in the investigated strain was monitored by PCR amplification assays using plasmid-specific primers.
2.7. Bacterial Growth Assay
Overnight cultures were diluted to an OD_600_ of 0.1 and the OD_600_ was measured at 37 °C under shaking in triplicate over 22 h using a TECAN MPlex microplate reader and iControl 2.0 software (Tecan Trading AG, Männedorf, Switzerland). Pathlength correction was not applied.
2.8. Western Blot Analysis
Whole cell lysates (WCL) of bacterial cultures grown to OD_600_ 1.5 (mid-logarithmic phase) were prepared by pelleting at 4000 x rcf. Resulting supernatant fractions were precipitated with 20% final concentration of ice-cold TCA according to standard protocols. Samples were analyzed on an SDS-PAGE under reducing conditions and transferred to PVDF. After blocking in 5% skimmed milk powder for 1 h, the primary antibodies (anti-DYKDDDDK Tag, Cell Signalling Cat. No. 14793S or Anti-His Tag, GenScript Cat. No. 25B6E11) were incubated overnight at 4 °C. HRP-coupled secondary antibodies (Biozol, Hamburg, Germany) were applied 1:20,000 for 1 h at room temperature and developed with Femto ECL (Thermo Fisher) using an INTAS Professional imaging system (INTAS Science Imaging, Göttingen, Germany).
2.9. Differentiation of Human-Monocyte-Derived Macrophages (hMDMs)
Primary human macrophages were derived from peripheral blood mononuclear cells (PBMCs) isolated from leukoreduction system cones using the SepMate™-50 system (StemCell Technologies, #85450) and Ficoll-Paque (GE Healthcare, #17144003) gradient according to manufacturer’s instructions. Monocytes were purified from the PBMC fraction using the EasySep CD14+ system (StemCell Technologies, #17858) according to manufacturer’s instructions and seeded in 12-well cell culture plates, at a density of 2 × 10^5^ cells/well, in RPMI1640 (Thermo Fisher Scientific, #72400054) supplemented with 10% (v/v) heat-inactivated FBS and 50 ng/mL recombinant human macrophage colony-stimulating factor (M-CSF) (StemCell Technologies, #78057). Macrophages were allowed to differentiate for 7 days and used for experiments on day 8. For infections, 2 × 10^5^ hMDMs were infected with freshly cultured bacteria at MOI10. After 2 h infection, the cells were washed with PBS and extracellular bacteria were killed with 15 µg/mL gentamycin (Thermo Fisher) for 30 min before the medium was changed to 1 µg/mL gentamycin for the remining total infection duration as indicated.
2.10. Infection of RAW 246.7 Cells
RAW 246.7 cells (=ATCC TIB71) were routinely cultured in RPMI1640 supplemented with 1X GlutaMaXX and 10% heat-inactivated FCS (Thermo Fisher). For infections, 1 × 10^5^ RAW 246.7 cells were seeded the day before in a 12-well plate and infected with freshly cultured bacteria at MOI25. After 2 h infection, the cells were washed with PBS and extracellular bacteria were killed with 15 µg/mL gentamycin (Thermo Fisher) for 30 min before the medium was changed to 1 µg/mL gentamycin for the remaining total infection duration as indicated.
2.11. Flow Cytometry Analysis
After infection, RAW 246.7 or hMDMs cells were washed three times with PBS and a live/dead stain was performed with FVS660 (Becton Dickinson, Temse, Belgium) for 15 min at 4 °C. Afterwards, cells were fixed for 15 min with ROTI^®^Histofix (Roth) at room temperature. Cells were permeabilized in 0.1% Triton-X for 15 min, blocked in 1% BSA and 22.25 mg/mL glycine for 1 h and primary antibody (BD Difco™ Salmonella O Antisera) was applied in 1:500 dilution for 1 h. Secondary antibody (Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488, Invitrogen) was applied for 1 h at room temperature. Cells were scraped and analyzed with an Attune NxT flow cytometer. Gates were set based on negative controls (non-infected for Ty21a+ve and empty vector for mRFP+ve, see Supplementary Figure S5). Statistical analysis was performed with GraphPad Prism software version 10.0.
2.12. Immunofluorescence
Raw 246.7 cells or hMDMs seeded onto coverslips were washed three times after infection and fixed for 15 min with ROTI^®^Histofix (Roth) at room temperature. Cells were permeabilized in 0.1% Triton-X for 15 min, blocked in 1% BSA and 22.25 mg/mL glycine for 1 h and primary antibody (BD Difco™ Salmonella O Antisera) was applied in 1:1000 dilution for 1 h. Secondary antibody (Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488, Invitrogen) was applied for 1 h at room temperature. The slides were mounted with ProLong™ Glass mounting medium with NucBlue DNA stain.
3. Results
We developed a Ty21a-based vaccine platform technology (JMU-SalVac-system) in which the antigen delivery plasmids (pSalVac-plasmid-series) are stabilized by a ΔtyrS/tyrS^+^ based balanced-lethal system (BLS). In order to test the feasibility of the JMU-SalVac-system, we first constructed several BLS-stabilized vaccine strains (see Table 1). These include strains with the CDS of mature CTB as model antigen in A-site (see Figure 1) for secretion via the hlyA-secretion system (abbreviation BLS-ActxB_B0), the mRFP-gene integrated into the B-site (see Figure 1) under control of the in vivo inducible promoter asr (BLS-A0_B_Pasr_RFP), the mRFP-gene integrated into the B-site and under control of in vivo inducible promoter pagC (BLS-A0_B_PpacC_mRFP) and a control strain with empty A- and B-sites (BLS-A0_B0). Further control strains were a Ty21a wildtype vaccine strain transformed with plasmid pSalVac A_ctxB__B0 KanR (Ty21a-ActxB_B0 KanR) and a BLS-stabilized strain with a kanamycin resistance gene retained on the pSalVac plasmid (BLS-ActxB_B0 KanR). The latter strain serves as a reporter strain for the plasmid retention status in colonies of the BLS strain. The kanamycin resistance gene of BLS-A_ctxB__B0 KanR is not needed for the retention of the plasmid but facilitates readout during the plasmid stability assay. To test stability of the ΔtyrS/tyrS^+^ BLS, both BLS-stabilized strains BLS-A_ctxB__B0 and BLS- A_ctxB__B0 KanR as well as the non-stabilized kanamycin-resistant strain Ty21a-A_ctxB__B0 KanR were tested for plasmid stability by serial passages of cultures at 37 °C in LB-medium for 6 days in the absence of antibiotic selection. For comparison, strain Ty21a harboring the conventional antigen expression plasmid pMKhly-CtxB [10] was also included. Plasmid maintenance in strains harboring plasmids with KanR genes was determined by examining resistance to kanamycin daily. The ratio of colonies growing on TS agar with kanamycin compared to TS agar without kanamycin was calculated. For the BLS-stabilized strain BLS-ActxB_B0, which does not carry an antibiotic resistance gene, a plasmid-specific PCR analysis was performed with at least 100 colonies from TS agar plated from serial dilutions of the starter culture (generation 0) and after 6 days of serial passaging. Plasmids without BLS stabilization showed low stability, as less than 80% of the picked colonies were able to grow on kanamycin containing TS-agar after just one day of serial passage (see Figure 3A). After 6 days, this fraction of kanamycin resistant colonies dropped to less than 3%. In contrast, both BLS-stabilized pSalVac plasmids showed 100% retention after 6 days of serial passage (for absolute numbers see Supplementary Table S2). No change in nucleotide sequence of the CTB-expressing A-site of BLS-ActxB_B0 was observed in 10 sequenced clones after 6 days of passage, and an mRFP-expressing BLS strain (BLS-ActxB_BDR) showed mRFP expression in 100% of tested colonies after the same 6-day passage duration.
Effective immunization with live vector vaccines depends on high and sustained antigen expression, for which plasmid stability is crucial. To simultaneously ensure the fitness of the vaccine strains, the pSalVac vector system enables the efficient secretion of soluble antigens via the hlyA secretion system, limiting the toxicity of difficult-to-express antigens. Antigens destined for secretion are cloned in the designated A-site to create a fusion protein with the hlyA N-terminus and the hlyA-signal fused N- and C-terminally, respectively. This enables expression via the constitutively active hlyA promoter and secretion of the protein as shown here for CTB-FLAG (Figure 3E). The CTB-FLAG signal in the supernatant does not originate from autolysis of the vaccine strains, as virtually none of the cytosolically expressed TyrS-His was detected in the supernatant. Expression of CTB did not significantly alter the growth behavior of the carrier strain, as shown by growth curve analysis (Figure 3C) and the corresponding area under the curve analysis of the growth curves (Figure 3B). Furthermore, expression of CTB-FLAG did not change after 6 days of serial passage (Figure 3F). In addition to the A-site, the so-called B-site, which is located downstream of the hlyA secretion apparatus genes, can be used for expression. We illustrate this in this study with in vivo inducible promoters such as the acid shock response promoter (P_asr_) and SPI-II promoter P_pagC_, which enable the regulated delayed antigen expression of otherwise toxic proteins with minimal loss of fitness for the carrier [2,46]. P_asr_- and P_pagC_-mRFP promoter fusions were cloned into the B-site of pSalVac and transformed into a BLS-stabilized vaccine strain. These strains were used to infect the macrophage cell line RAW246.7 (Figure 4). Microscopical and flow cytometry analysis of infected cells shows that mRFP is expressed intracellularly in up to ~30% of all macrophages, with P_asr_-mRFP in above 50% of infected macrophages (see Figure 4B right panel).
To demonstrate the expression of model antigens by P_asr_ and P_pagC_ in a model relevant to vaccination of humans, human-monocyte-derived macrophages (hMDMs) were infected with the BLS-stabilized Ty21a reporter strains. Both promoters (Figure 5) were induced in infected primary hMDMs, indicating that intracellular vaccine strains encounter an environment permissive for promoter induction.
Administration of a combination of multiple antigens may prevent immune escape of different serovars, but the simultaneous expression of different antigens from the same vaccine host may increase metabolic burden and reduce fitness. Using both expression cassettes for expression of secretory and cytosolic antigens (CTB-FLAG and mRFP), we tested invasion and intracellular antigen expression in hMDMs. To monitor intracellular replication of the strains, a fluorescence reporter cassette on pFCcGi with constitutive mRFP and arabinose-inducible GFP expression was cloned into the B-site of ActxB_B0. Since GFP expression is only induced before the infection, the signal is diluted upon proliferation of Salmonella after infection and only the RFP fluorescence is retained. Non-proliferating bacteria retain both fluorescence signals. The BLS-stabilized strain expressed CTB-FLAG with and without induction of GFP via arabinose (Figure 6A) but showed a slight reduction in replication as shown by growth curve measurements (Figure 6B). Nevertheless, BLS-ActxB_BDR proliferated in the hMDM infection model, as revealed by varying GFP signal strength in mRFP-positive bacteria in the hMDMs (Figure 6C). While live cell imaging also showed intracellular replication in hMDM on the single cell level, bacterial fluorescence decreased across the whole field of view during the measured time course (see Supplementary Movie S1 and Figure S9). These data indicate that the BLS-stabilized carrier strain expressing heterologous proteins in A- and B-site still has sufficient fitness to infect and survive in the hMDM model for a substantial period.
4. Discussion
We developed a plasmid system for antigen expression and delivery for Salmonella enterica serovar Typhi strain Ty21a (Ty21a), the approved attenuated bacterial vaccine against typhoid fever. This vaccine strain has been administered to more than 200 million recipients worldwide over 25 years and has never become virulent again [47], which emphasizes its excellent safety profile. The effect of pre-existing immunity to Salmonella carrier strains on the response to heterologous antigens has been investigated in several studies with mixed results, some showing an enhanced immune response [48,49,50] and others a reduction [51,52] (reviewed in [53]). Most relevant to the present work, studies on Ty21a showed no effect [54] or even an enhancement of the immune response against the heterologous antigen in carrier-primed human vaccinees [49]. The authors speculate that the choice of antigen is an important factor in determining the outcome of vaccination in such individuals [49]. Although Ty21a has been established as a versatile oral vaccine vector for expression of various foreign antigens [55], there is currently no stable, non-auxotrophic, non-antibiotic resistance plasmid system available for antigen expression in Ty21a. The JMU-SalVac-system described in this study closes this important gap.
As the use of antibiotics or other external treatments for plasmid stabilization in vaccinees is not possible, other selection mechanisms had to be developed to ensure retention of the expression plasmids (for recent review about Salmonella-based carrier vaccine strains, see [22]. This has previously been achieved in *Salmonella-*based vaccine strains, e.g., by the knockout of the aspartate β-semialdehyde dehydrogenase (Asd) and in trans complementation on the expression plasmid [56]. Stabilization after chromosomal deletion of asd results in DAP auxotrophy. However, plasmids stabilized by in trans complementation of auxotrophies may be lost if, e.g., auxotrophies in essential metabolic pathways are complemented by environmental metabolites.
We used a different strategy here by deleting the essential tyrS gene in the genome of Ty21a and complementation of tyrS on a low to medium copy vector, resulting in a non-antibiotic and non-auxotrophic stabilization. Our data show that the ΔtyrS/tyrS^+^ based JMU-SalVac-system provides stability of the plasmid without further selection, whereas non-stabilized plasmids were rapidly lost under the same conditions. Since the tyrS gene is essential and functional complementation by environmental metabolites is excluded [26,57], the stability of the JMU-SalVac system should also be given in complex in vivo situations. Furthermore, in trans complementation of tyrS had no negative impact on the fitness of the vaccine strain, determined by growth and infection behavior. This is of crucial importance as Ty21a is already strongly attenuated by mutations in several virulence genes, which makes it a good candidate for safe mass vaccination from the outset. In addition, the expression of model antigens remained stable during the tested 6-day passage of the strains, which is important for a long-lasting production of antigens during the vaccination phase (see Figure 3). Our system overcomes the challenges of previous approaches to non-auxotrophic stabilization. SSB-based stabilization has shown expression instability after several days of passage, which is not seen with the TyrS-based BLS, as evidenced by sustained resistance to kanamycin in the BLS-ActxB_B0 KanR strain, mRFP expression in all colonies of BLS-ActxB_BDR and unchanged expression of CTB-FLAG after 6 days of passage (Figure 3F) [24]. Higher copy number plasmids, which may be advantageous for high antigen expression, were also no problem for plasmid stability with the TyrS-stabilized vectors as opposed to stabilization with SSB [24]. In addition, this is the first such system described for the Ty21a vaccine strain.
Important functional features of the pSalVac plasmids are two different sites for expression and delivery of antigen proteins. The first expression cassette (A-site) includes the hly operon and two short residual sequences of the hlyA N-terminus and the C-terminal signal sequence (hlyAs) for recognition and secretion via the hemolysin secretion system [9,40]. One advantage of using the hlyA system for secretion of antigens into the extracellular space is that a relatively short secretion signal of the hlyA toxin has to be fused to the antigen for efficient secretion. In this study we use CTB as model antigen and show efficient secretion into the culture supernatant. CTB was selected here since it has very strong adjuvant activity, particularly for the induction of mucosal immunity [58,59]. Protein antigens fused to CTB are stabilized and CTB directs the antigen fusion protein to its receptor ganglioside GM1 for uptake by antigen-presenting cells.
The second expression cassette (B-site) is located outside the hly gene cluster and is variable with respect to promoter, signal peptide and thus also the expression strength and the subcellular localization of the fusion protein. The simultaneous expression of different antigens, which was shown here with CTB-FLAG (A-site) and mRFP (B-site), can be advantageous to avoid immune escape of pathogens. The two different expression cassettes thus offer a high degree of flexibility, e.g., with regard to the expression and subcellular localization of vaccine antigens, and represent an alternative to published dual plasmid systems [20].
Oral vaccination with encapsulated lyophilized Ty21a ensures the successful passage of the bacteria through the gastric acid barrier [60]. In the small intestine, the bacteria invade the mucosa and translocate to the intestinal lymphoid follicles and the draining mesenteric lymph nodes, where they interact with mononuclear phagocytic cells of the lymphoid follicles. We therefore tested infection of RAW246.7, and particularly human-monocyte-derived macrophages (hMDMs) as relevant target cells for Ty21a BLS strains. In contrast to the infection experiments with P_asr_-mRFP reporter fusions performed by others [43], in which Ty21a was unable to induce active infection of THP-1 derived macrophages, the strains constructed here with a similar reporter fusion infected and in some cases even multiplied in macrophages as was measured with a dual reporter system and live cell imaging (see Figure 6 and Supplementary Movie S1) [34]. As expected, bacterial fluorescence decreased during the time course of live cell imaging however (Supplementary Figure S9), which indicates that the majority of internalized Ty21a is degraded by the hMDM cells. The eventual degradation of the vaccine host is important for the delivery of non-secreted antigens as was modelled here with mRFP. The use of in vivo inducible antigen expression under the control of the acid-inducible P_asr_ and the Mg^2+^-sensing P_pagC_ revealed strong expression in the majority of cells. Infected cells without mRFP expression likely had not yet acidified (in the case of the asr construct) or depleted the Mg^2+^ concentration enough for the reporter to be visible in flow cytometry at the time of measurement. Such in vivo-inducible promoters can facilitate the immunization with difficult to express antigens, limit the metabolic burden of antigen expression, and can have favorable effects on vaccination outcome, possibly due to increased fitness of the strongly attenuated carrier strain [22].
The next step is to validate the developed expression system in a suitable in vivo model. However, even positive immunization results obtained in mice or other animals are not indicative of an effective vaccine, as Ty21a cannot infect animals orally. Since S. Typhi is an exclusively human pathogen, clinical studies are the most important next step for the validation of antigen expression systems [61]. This will show if the non-auxotrophic approach here is an improvement over previously demonstrated BLSs that failed to induce protection with Ty21a in the clinical setting [62,63].
In summary, we have constructed and tested a novel antibiotic resistance gene-free, non-auxotrophic plasmid stabilization system for Ty21a-based live vaccines in which antigens can be stably, safely, and efficiently expressed. Bacterial strains developed with our system have the potential to be used in humans as safe and stable vaccines against mucosal infections.
5. Patents
A patent covering the construction and use of pSalVac vectors has been filed (WO 2022/034221 A1).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Clark-Curtiss J.E. Curtiss R. Salmonella Vaccines: Conduits for Protective Antigens J. Immunol.2018200394810.4049/jimmunol.160060829255088 · doi ↗ · pubmed ↗
- 2Bumann D. Regulated antigen expression in live recombinant Salmonella enterica serovar Typhimurium strongly affects colonization capabilities and specific CD 4(+)-T-cell responses Infect. Immun.2001697493750010.1128/IAI.69.12.7493-7500.200111705925 PMC 98839 · doi ↗ · pubmed ↗
- 3Galen J.E. Curtiss R. The delicate balance in genetically engineering live vaccines Vaccine 2014324376438510.1016/j.vaccine.2013.12.02624370705 PMC 4069233 · doi ↗ · pubmed ↗
- 4Kang H.Y. Curtiss R. Immune responses dependent on antigen location in recombinant attenuated Salmonella typhimurium vaccines following oral immunization FEMS Immunol. Med. Microbiol.2003379910410.1016/S 0928-8244(03)00063-412832112 · doi ↗ · pubmed ↗
- 5Galen J.E. Zhao L. Chinchilla M. Wang J.Y. Pasetti M.F. Green J. Levine M.M. Adaptation of the endogenous Salmonella enterica serovar Typhi cly A-encoded hemolysin for antigen export enhances the immunogenicity of anthrax protective antigen domain 4 expressed by the attenuated live-vector vaccine strain CVD 908-htr A Infect. Immun.2004727096710610.1128/iai.72.12.7096-7106.200415557633 PMC 529119 · doi ↗ · pubmed ↗
- 6Gentschev I. Dietrich G. Spreng S. Neuhaus B. Maier E. Benz R. Goebel W. Fensterle J. Rapp U.R. Use of the alpha-hemolysin secretion system of Escherichia coli for antigen delivery in the Salmonella typhi Ty 21a vaccine strain Int. J. Med. Microbiol. IJMM 200429436337110.1016/j.ijmm.2004.07.01015595386 · doi ↗ · pubmed ↗
- 7Hotz C. Fensterle J. Goebel W. Meyer S.R. Kirchgraber G. Heisig M. Fürer A. Dietrich G. Rapp U.R. Gentschev I. Improvement of the live vaccine strain Salmonella enterica serovar Typhi Ty 21a for antigen delivery via the hemolysin secretion system of Escherichia coli Int. J. Med. Microbiol. IJMM 200929910911910.1016/j.ijmm.2008.06.00618706861 · doi ↗ · pubmed ↗
- 8Gentschev I. Dietrich G. Goebel W. The E. coli alpha-hemolysin secretion system and its use in vaccine development Trends Microbiol.200210394510.1016/s 0966-842x(01)02259-411755084 · doi ↗ · pubmed ↗