The Development of Visible-Light Organic Photocatalysts for Atom Transfer Radical Polymerization via Conjugation Extension
Hui Shao, Runzhi Long, Hui Xu, Pan Sun, Guangrong Wang, Yuanming Li, Saihu Liao

TL;DR
Researchers developed new organic photocatalysts that work under visible light to control polymerization reactions efficiently.
Contribution
A new class of visible-light-responsive organic photocatalysts was developed for efficient polymer synthesis.
Findings
Annulated N-aryl benzo[kl]acridines were found to be effective visible light photocatalysts.
Selenium-doped structures enabled controlled polymerization of methacrylates with low catalyst loading.
The method produced polymers with controlled molecular weights and low dispersities.
Abstract
This work aimed to develop organic photocatalysts (PCs) that could mediate organocatalytic atom transfer radical polymerization (O-ATRP) under visible light. Through the core-modification of known chromophoric structures and ring-locking to reach a conjugation extension, annulated N-aryl benzo[kl]acridines were identified as effective visible light-responsive photocatalysts. The corresponding selenium-doped structure showed excellent performance in the O-ATRP of methacrylates, which could afford polymer products with controlled molecular weights and low dispersities under the irradiation of visible light at a 100 ppm catalyst loading.
Click any figure to enlarge with its caption.
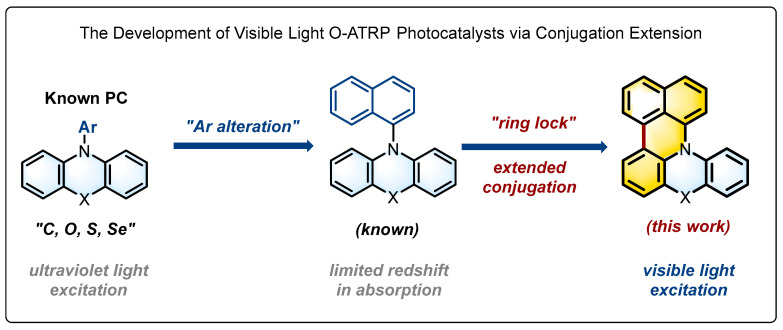 Figure 1
Figure 1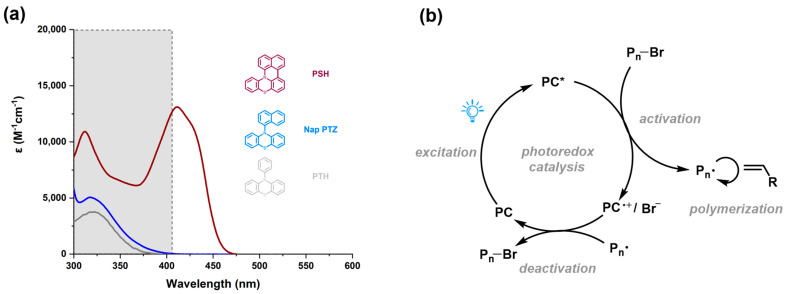 Figure 2
Figure 2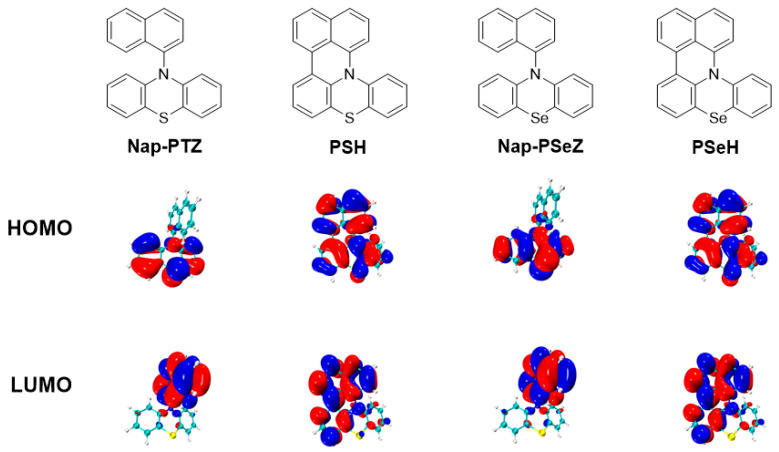 Figure 3
Figure 3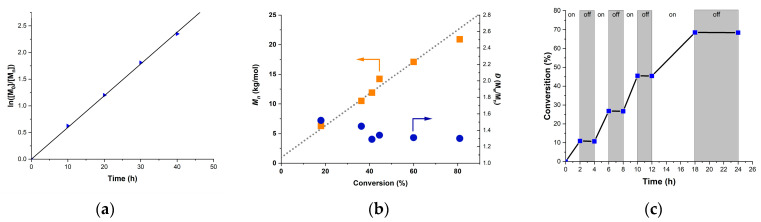 Figure 4
Figure 4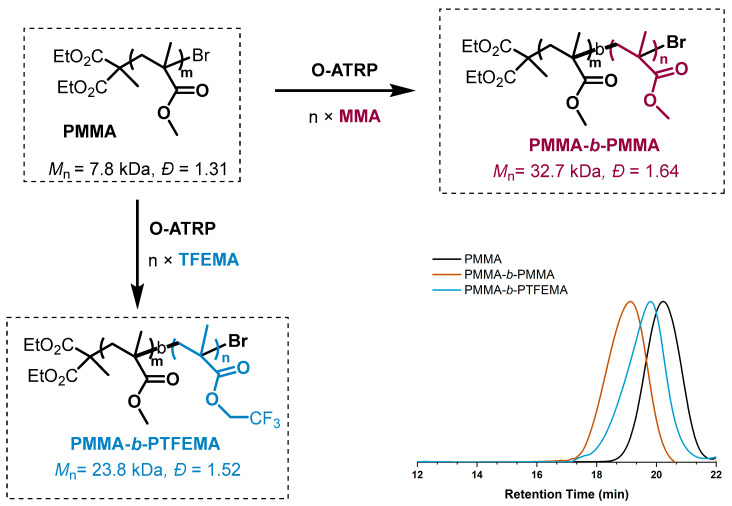 Figure 5
Figure 5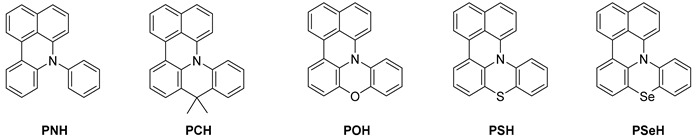 Figure 6
Figure 6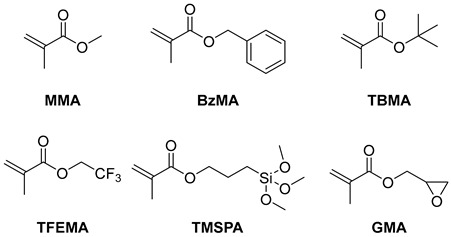 Figure 7
Figure 7- —National Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced Polymer Synthesis and Characterization · Photopolymerization techniques and applications · Radical Photochemical Reactions
1. Introduction
Reversible deactivating radical polymerization (RDRP) is a polymerization technology that has been rapidly developing in the past three decades and has found wide applications in many fields [1,2,3]. The application of light regulation in RDRP-based polymer synthesis has been an emerging and rapidly growing research area over the last ten years [4]. The advantage of light as an external stimulus to regulate the polymerization process includes the non-invasive mediation, high energy efficiency, spatiotemporal control, wide availability, and easy access to inexpensive, long-wavelength LED light sources [3]. In particular, the corresponding visible light-mediated RDRP offers photocontrol technologies enabling temporal and spatial regulation of chain growth, a capability not attainable in conventional thermally activated and chemically activated processes [4,5]. As one of the most powerful and versatile RDRP techniques, atom transfer radical polymerization (ATRP) [6,7] has gained significant advances in the past years by uniting light regulation, and the corresponding photocontrolled ATRP (photo-ATRP) routes not only allow for the precise control of reaction kinetics but also facilitate the fabrication of advanced materials [8,9,10].
Light-mediated radical polymerization has profited from the rapid development of organic photocatalysts (PCs). In this regard, the development of photocatalysts derived from phenothiazine [11,12], phenazine [13], and phenoxazine [14,15] core structures (Figure 1, left) with favorable redox balance between reducing excited states and oxidizing radical cations to mediate the activation–deactivation equilibrium [16] has significantly pushed forward the field of O-ATRP. To further increase the performance of these photocatalysts, various modifications have been introduced to the core structures, aiming to enhance their absorption of visible light [16] and enable O-ATRP with good control under longer wavelength light rather than ultraviolet [17,18,19,20,21]. On the other hand, variation in the heteroatoms in these structures has also been investigated, including systematic studies on the effect of chalcogenide atoms [22,23], which not only improved our understanding of the structure–property relationships but also offered a tentative guideline for tailoring the structure of known PCs.
Along with the modifications of known chromophoric structures like phenothiazine, our research interest has moved toward developing photocatalysts with distinct parental skeletons. In this regard, we became particularly interested in the polycyclic arenes with a conjugated structure larger than that of anthracene as a parental framework for “doping”, following the design logic of heteroatom doping of polycyclic arenes for the development of new photocatalyst [23,24,25,26]. Herein, we wish to report that, following additional structural refinement, N-naphthyl phenochalcogenazine analogue (Figure 1, middle) could evolve into a visible-light photocatalyst, with its absorption maximum redshifting to above 400 nm after “locking” the naphthyl group, which could thus serve as a visible-light photocatalyst for the O-ATRP of methacrylates. These locked structures possess an extended conjugation (Figure 1, right), with the benzo[kl]acridine moiety (highlighted in yellow) exhibiting a strong absorption of purple-blue light. Upon further variation in the doping atoms (X = C, O, S, and Se), the corresponding selenium-doped structure was found showing the best performance in the O-ATRP of methacrylates under the irradiation of visible light, allowing for the synthesis of polymers with controlled molecular weights and low dispersities.
2. Results and Discussion
2.1. Photocatalyst Development
Molecular Design and Synthesis. To increase the visible-light absorption of the phenothiazine and phenoxazine chromophoric structures, core-modification via alteration of the Ar group (e.g., replacing the phenyl group with a naphthyl group) has been commonly employed, but the improvement like redshift of absorption was often limited [27,28]. To generate a structure with an extended conjugation, we conceived the combination of Ar alteration with “ring lock” could lead to a new organic photocatalyst for O-ATRP that could probably be effective under visible light (Figure 1). We thus synthesized five photocatalysts with a benzo[kl]acridine moiety, by varying the exo-doping atoms (X = C, O, S, Se, shown below). The detailed synthesis is described in the Supporting Information. These structures can be readily prepared from phenochalcogenazines through sequential N–H/C–H coupling with 1,8-dibromonaphthalene in a sequential transformation [29,30,31].
Properties of the catalysts: The UV-Vis absorbance of each PC was first measured, and the results are summarized in Table 1. The corresponding UV-Vis absorption spectra are in Supplementary Figure S12. All five photocatalysts exhibited a strong absorption in the range of 350–450 nm, with the maximum absorption wavelength located at 408–435 nm. A comparison of the absorption profiles of PSH with known photocatalyst N-naphthyl phenothiazine (Nap-PTZ) and N-phenyl phenothiazine (PTH) is depicted in Figure 2a. Replacing the N-phenyl group in PTH with a larger aryl group like naphthyl was commonly used to enhance the catalysts’ absorption of visible light, but the improvement was often limited. In contrast, the absorption profile of PSH was not only redshifted (Δλ_max_ = 95 nm versus Nap-PTZ) into the visible-light region (λ_max_ = 412 nm), and it also exhibited a significantly enhanced molar extinction coefficient (ε = 13,111 M^−1^cm^−1^ at λ_max_ = 412 nm), making it more efficient in absorbing visible light than the unlocked structure (Nap-PTZ). In fact, all five photocatalysts exhibited a strong visible-light absorption with molar extinction coefficients of ε_max_ > 10,000 M^−1^ cm^−1^, which could favor their excitation by longer wavelength light during the photocatalytic polymerization (Figure 2b) and thus decrease the catalyst loadings [32,33].
Cyclic voltammogram (CV) measurements were also performed with these catalysts, and all the five catalysts, including PSH and PSeH, showed a reversible CV, suggesting the corresponding radical cations’ good stability, which is critical in the deactivation step to convert the propagating radicals (P_n_·) into dormant chains. Based on the E1/2(PC^•+^/PC) values obtained from CV measurements and the λ_em_ of fluorescence emission, the reduction power of the excited states (^1^PC*) of these catalysts can be calculated. As shown in Table 1, all of these catalysts possess a good reducing ability at the excited states (−2.00 to −2.25 V vs. SCE) that is enough to reduce the alkyl bromide initiators via single-electron transfer [11,12]. Moreover, DFT calculations were performed to evaluate the reduction capability of the triplet states [34,35,36,37,38], and the E^0^*** values (PC^•+^/^3^PC*) are summarized in Table 1, in the range from −1.43 to −1.78 V vs. SCE. The E^0^*** values (PC^•+^/^3^PC*) obtained with our method for PCH and PSeH (−1.78 V and −2.04 V vs. SCE) show good agreement with the experimental values (−2.25 V and −2.03 V vs. SCE). In addition, both Eºexp(PC^•+^/^1^PC*) and Eºtheo(PC^•+^/^3^PC^*^) follow similar trends to those seen in the excited-state energies (i.e., decreasing in magnitude from POH to PSH, and then increasing from PSH to PSeH) (Supplementary Table S1). Furthermore, heavy-atom (e.g., Se) doping can often enhance the spin–orbit coupling (SOC) and favor the intersystem crossing from singlet states into triplet states, which could often improve the initiation and activation in O-ATRP [39,40].
On the basis of time-dependent density functional theory (TD-DFT) calculations at the B3LYP/6-311+G (d)/LANL2DZ level of theory, the HOMOs and LUMOs are mainly located on the donor and acceptor groups, respectively (HOMO = highest occupied molecular orbital; LUMO = lowest unoccupied molecular orbital) (Figure 3). Among these PC molecules, PSH and PTZ are similar in that they have one phenothiazine donor attached to the naphthalene ring, but with a different conjugation due to the lock or not of the naphthalene ring. Comparing PSH and PTZ, we see that the locked structure has a large contribution to both HOMO and LUMO density, and the HOMO and LUMO overlap is extensive when the naphthalene ring is locked. Therefore, the molecular orbital picture of PSH suggests an obvious expanded conjugation. Furthermore, an examination of the triplet state (^3^PC*) frontier orbitals also reveals differences between locked-structure PCs and the unlocked ones (Supplementary Figure S17). The low-lying singly occupied molecular orbital (SOMO) of the PSeH differs from Nap-PSeZ, of which the electron is mainly localized over the phenothiazine π system. This molecular orbital picture also indicates an obvious extended conjugation in these locked structures.
2.2. Catalyst Evaluation
The catalytic performance of PCs was initially evaluated in the polymerization of methyl methacrylate (MMA) in dichloromethane (DCM) by using diethyl 2-bromo-2-methylmalonate (DBMM) as an initiator at a [MMA]0/[DBMM]0/[PC]0 ratio of 1000/10/0.1. As shown in Table 2, PNH afforded a poly(MMA) product with a high Đ (1.49) and a number average molecular weight that deviates significantly from the corresponding theoretical values (entry 1). POH exhibited a better catalytic performance in the O-ATRP of MMA when compared to PNH, producing PMMA with a similar dispersity but a much higher initiator efficiency (I* = 93%, entry 2). The introduction of the S atom also gave a higher I* (74%, entry 4) in comparison to PNH (entry 1) and PCH (entry 3). Entries 5 and 6 in Table 2 are the results of polymerizations under ultraviolet (UV) light for 8 h. In this case, PSH could also afford MMA polymer products with lower dispersity. To our delight, among these catalysts, the corresponding selenium analogue (PSeH) showed the best performance in terms of molecular-weight control (I* > 85%) and low dispersity (Đ 1.24 at >80% conversion), and both purple and white LEDs are suitable to achieve a controlled radical polymerization (entries 7 and 8). In contrast, the unlocked one (Nap-PSeZ) gave a less controlled polymerization and a much lower initiator efficiency (entry 9).
2.3. Kinetic Study, Temporal Control, and Polymerization Extension
The kinetics of the polymerization of MMA were studied at catalyst loadings of 100 ppm, as shown in Figure 4a,b, which unveiled a linear growth of molecular mass upon monomer conversion (up to 80% conversion). It can be observed that there is a linear relationship in the whole process of polymerization, and the dispersity remains within a relatively narrow range, demonstrating a constant concentration of propagating radicals and a pseudo first-order kinetics of the photo-ATRP. Effective temporal control over the polymerization by irradiation was also demonstrated through the light on–off experiment (Figure 4c), and no monomer conversion was observed during the dark periods. It can be assumed that photoexcited PCs activate the dormant species, and the chain grows only when the light is on, thus achieving a light regulation over the chain propagation.
PSeH was further applied to the polymerization of different monomers, and the results are summarized in Table 3. The polymerization of methacrylate monomers, including benzyl methacrylate (BzMA), trifluoroethyl methacrylate (TFEMA), tert-butyl methacrylate (t-BMA), and 3-(trimethoxysilyl)propyl methacrylate (TSPMA), proceeded in a controlled manner in terms of molecular weight and low dispersity under white LEDs (entries 1–5). It is worth noting that the PSeH could also deliver an efficient polymerization of glycerol acrylate (GMA). Furthermore, chain-extension polymerizations were also carried out using the isolated PMMA macroinitiator (Mn = 7.8, Đ = 1.31), which was prepared with PSeH as the photocatalyst and DBMM as the initiator under the standard reaction conditions. As shown in Figure 5, two block polymers, PMMA-b-PMMA and PMMA-b-PTEFMA, were successfully prepared, indicating that good fidelity of the chain-end groups was achieved in the PSeH-mediated radical polymerization.
3. Materials and Methods
3.1. Materials and Instruments
The monomers (methyl methacrylate (MMA), 2,2,2-trifluoroethyl methacrylate (TFEMA), 3-(trimethoxysilyl)propyl methacrylate (TMSPA), benzyl acrylate (BzA), and glycidyl methacrylate (GMA)) were dried over calcium hydride, distilled, and stored under an inert atmosphere at 2–8 °C. Diethyl 2-bromo-2-methylmalonate (DBMM) was purchased from TCI Chemicals and dried over calcium hydride, distilled, and stored under an inert atmosphere at 2–8 °C. Dichloromethane (DCM), tetrahydrofuran (THF), and toluene were dried over calcium hydride, distilled, and stored under an inert atmosphere at 2–8 °C. Other solvents, N,N-dimethylacetamide (DMAc), N,N-dimethylformamide (DMF), and ethyl acetate (EtOAc), were purchased from J&K Seal and used as received.
^1^H NMR and ^13^C NMR spectra were recorded on a Bruker AVIII 400 or a Bruker Avance 500 spectrometer (Bruker, Billerica, MA, USA), using dimethylsulfuxide-d_6_ (DMSO-d_6_) or CDCl_3_ as the solvent. Analysis of polymer samples was performed via gel permeation chromatography (GPC), using an Agilent HPLC (Agilent Technologies Inc., Santa Clara, CA, USA), two Shodex GPC KD-806M gel permeation columns (8.0 mm ID × 300 mm), and a Wyatt Technology TrEX differential refractometer (Wyatt Technology, Santa Barbara, CA, USA), using THF as the eluent at a flow rate of 1.0 mL/min. The GPC system was calibrated based on narrow molecular distributions of poly(methyl methacrylate) standards with molecular weights between 602 and 2,200,000 g·mol^−1^. Cyclic voltammogram (CV) measurement experiment was carried out with a CHI660 D electrochemical workstation (Shanghai Chenhua Instrument Plant, Shanghai, China), with 0.1 mol of tetrabutylammonium hexafluorophosphate as the electrolyte at room temperature, and an Ag/AgCl electrode was used as the reference electrode. UV/Vis/NIR spectra were recorded on a Perkins Elmer Lambda 900 spectrometer (PerkinElmer Life and Analytical Sciences, Shelton, CT, USA) equipped with a PTP-1 Peltier temperature controller, and steady-state emission spectra were acquired using Edinburgh Instruments, FLS980 spectrometer (Edinburgh Instruments, Livingston, UK). Mass spectra were recorded on an Agilent Q-TOF 6520 system (Agilent Technologies Inc., Santa Clara, CA, USA) using electrospray ionization in Positive/Negative ion detection (ESI^+^/ESI^−^) mode, and the significant fragments are reported in the following fashion: m/z (relative intensity).
3.2. Synthesis and Characterization of Catalyst PSeH
Synthesis of PSeH precursor RM 5: Bis (2-iodophenyl) amine (1.26 g, 3 mmol, 1 eq.), Se powder (0.474 g, 6 mmol, 2eq.), and KOH (0.672 g, 12 mmol, 4 eq.) were dissolved in 30 mL of DMSO. The reaction mixture was stirred at 110 °C for 24 h. At room temperature, the reaction mixture was diluted with saturated NH_4_Cl aq. solution and DCM. The aqueous phase was extracted by DCM three times and dried with Na_2_SO_4_, and the solvent was removed by rotary evaporation. The crude product was purified by flash chromatography, with a yield of 75%.
Then, 1,8-dibromonaphthalene (3.5 mmol, 1 g), the abovementioned obtained precursor RM 5 (3 mmol), NaO*^t^*Bu (7 mmol, 0.67 g), Pd (OAc)2 (3 mol%, 0.09 mmol, 0.02 g), Pd_2_(dba)3 (3 mol%, 0.09 mmol, 0.082 g), Cy_3_P (7 mol%, 0.21 mmol, 0.06 g), and (tBu)3_P (7 mol%, 0.21mmol, 0.043 g) were stirred in 50 mL of dried toluene at 90 °C for 10 h. Then, the solvent was removed under reduced pressure, and the residue was purified by column chromatography, using hexane/dichloromethane as eluent on silica. Finally, the target products were obtained in 45–78% yield. Further recrystallization was conducted using hexane and DCM. PSeH ^1^H NMR ((500 MHz, CDCl_3) δ 7.73 (t, J = 7.6 Hz, 2H), 7.63 (d, J = 8.2 Hz, 1H), 7.49–7.44 (m, 1H), 7.40 (d, J = 8.2 Hz, 2H), 7.35–7.30 (m, 2H), 7.21 (d, J = 7.6 Hz, 1H), 7.13–7.07 (m, 2H), 7.05 (td, J = 7.5, 1.0 Hz, 2H).) ^13^C NMR (126 MHz, CDCl_3_) δ 144.4, 140.4, 138.1, 134.2, 131.3, 129.8, 129.2, 127.9, 127.2, 126.6, 126.2, 125.9, 125.3, 125.0, 124.3, 122.5, 121.5, 120.7, 120.4, 116.2, 114.1. HRMS (m/z): calcd for [M + H]^+^ C_22_H_14_NSe, 372.0286; found 372.0271. For characterization (NMR spectrum, UV-Vis, emission spectrum, lifetimes, and CV spectrum), please see the Supporting Information.
3.3. Typical Procedures for Photoinduced O-ATRP
A typical O-ATRP procedure under the standard reaction conditions is as follows: in the glovebox, MMA (0.5 mL, 4.7 mmol, 100 eq.) as the model monomer, DBMM (9 μL, 47 μmol, 1 eq.), PSeH (0.35 mg, 0.94 μmol, 0.02 eq), and 0.5 mL of DCM were successively added to the Schlenk tube. Subsequently, the tube was placed under the irradiation of purple or white LEDs for a specified reaction time. Then, the tube was opened under argon, and a 20 μL of reaction mixture was syringed out and added to CDCl_3_ (containing 250 ppm BHT) for ^1^H NMR analysis to determine the monomer conversion. After polymerization, the reaction mixture was poured into methanol (30 mL), and the precipitates were collected and dried to afford the purified polymer for further analysis by GPC.
3.4. Polymerization Procedure for Chain Extension from PMMA Macroinitiator
MMA (2.00 mL, 18.8 mmol, 100 eq.), DBMM (72 μL, 283 μmol, 1.5 eq.), and catalyst (0.188 μmol, 0.01 eq.) were dissolved in 2.50 mL DCM and reacted according to the above general polymerization procedure for 16 h. After that, the tube was opened under argon, and 20.0 μL of the mixture was syringed out and quenched into CDCl_3_ containing 250 ppm BHT to determine the monomer conversion by ^1^H NMR (Conv. = 70.4%). At this time, the reaction mixture was poured into 250 mL methanol and stirred for 4 h. The resulting precipitate was slowly dripped into room-temperature methanol, after stirring for half an hour, and then isolated by vacuum filtration and washed with excess methanol. The polymer was then re-dissolved in a minimal amount of DCM again and dripped into 150 mL of methanol and stirred for 2 h to fully remove unreacted monomer, initiator, or catalyst. The resulting PMMA was collected via vacuum filtration and dried in a vacuum, at 40 °C, overnight, and obtained 1g of polymer, Mn = 7.8 kDa, Ɖ = 1.31.
PMMA-b-PMMA: A Schlenk tube with a PTFE stirring bar was charged with 0.35 mg of PSeH (9.42 × 10^−4^ mol, 0.1 eq.) and 74 mg of the PMMA macroinitiator described above (Mn = 7.8 kDa, 1.0 eq.) that was dissolved in 1 mL of DCM. Then, 200 μL of MMA was added (0.19 mmol, 200 eq.) and reacted according to the above general polymerization procedure for 16 h. Then, the reaction mixture was loaded into a syringe and slowly dripped into room-temperature methanol to precipitate the polymer. The product was re-dissolved in HPLC-grade, unstabilized tetrahydrofuran, and the Mn and Ɖ were analyzed by GPC.
PMMA-b-PTFEMA: A Schlenk tube with a PTFE stirring bar was charged with 50 μL of PC (9.42 × 10^−4^ mol, 0.05 eq.) and 74 mg of the PMMA macroinitiator described above (Mn = 7.8 kDa, 1.0 eq.) which were dissolved in 1 mL of DCM. Then, 132 μL of TFEMA was added (1.12 mmol, 124 eq.) and reacted according to the above general polymerization procedure for 9 h. Then the reaction mixture as loaded into a syringe and slowly dripped into room temperature methanol to precipitate the polymer, The product was re-dissolved in HPLC grade, unstabilized tetrahydrofuran and the Mn and Ɖ were analyzed by GPC.
3.5. General Methods for Analysis of Kinetics and Molecular Weight Growth
A typical procedure of kinetics experiments were performed in glovebox, using a [MMA]:[DBMM] ratio of 200:1, with 100 ppm PSeH and 1 mL:1.5 mL MMA:DCM. To evaluate the kinetics and growth of molecular weight versus conversion for polymerization, an aliquot of 0.05 mL of reaction mixture was taken and injected into a solution of CDCl_3_ containing 250 ppm of the radical inhibitor (BHT) at predetermined times after the start of the polymerization, as indicated (when the reaction mixture was exposed to light). After NMR analysis, the sample was subjected to GPC analysis to determine the Mn and Mw/Mn values.
4. Conclusions
Based on the idea of “locking” the N-naphthyl group in the N-naphthyl phenothiazine, a series of new photocatalysts with extended conjugation were forged which exhibited strong absorption in the visible-light region (λ_max_ = 412 nm), with a significant redshift (Δλ_max_ = 95 nm) from ultraviolet to purple-blue light. This distinct feature, in combination with suitable redox potentials, allows the corresponding O-, S-, and Se-doped photocatalysts to mediate the atom transfer radical polymerization of MMA under the irradiation of visible light rather than UV light. Notably, the selenium photocatalyst (PSeH) showed an excellent performance and a high initiating efficiency, which could deliver the polymer products with well-controlled molecular weights and low dispersities (Đ as low as 1.24) at a 100 ppm catalyst loading only. Furthermore, chain-extension and light on/off experiments were also performed, suggesting a good chain-end fidelity and good temporal control over the chain growth. In conclusion, the conjugation extension is demonstrated as a viable approach and also important for designing/modifying photocatalysts for O-ATRP, with the aim to improve the absorption of visible light and catalytic activity. We anticipate that these new photocatalysts could find further application in both small-molecule and macromolecule synthesis in the future.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Corrigan N. Jung K. Moad G. Hawker C.J. Matyjaszewski K. Boyer C. Reversible-Deactivation Radical Polymerization (Controlled/Living Radical Polymerization): From Discovery to Materials Design and Applications Prog. Polym. Sci.202011110131110.1016/j.progpolymsci.2020.101311 · doi ↗
- 2Corrigan N. Yeow J. Judzewitsch P. Xu J. Boyer C. Seeing the Light: Advancing Materials Chemistry through Photopolymerization Angew. Chem. Int. Ed.2019585170518910.1002/anie.20180547330066456 · doi ↗ · pubmed ↗
- 3Walsh D.J. Hyatt M.G. Miller S.A. Guironnet D. Recent Trends in Catalytic Polymerizations ACS Catal.20199111531118810.1021/acscatal.9b 03226 · doi ↗
- 4de Ávila Gonçalves S. Rodrigues P.R. Pioli Vieira R. Metal-Free Organocatalyzed Atom Transfer Radical Polymerization: Synthesis, Applications, and Future Perspectives Macromol. Rapid Commun.202142210022110.1002/marc.20210022134223686 · doi ↗ · pubmed ↗
- 5Huang Y.-S. Ejeta D.D. Kuo S.-W. Nakamura Y. Huang C.-F. Combinations (Є) among Controlled/Living Polymerizations and Utilizations of Efficient Chemical Reactions for the Synthesis of Novel Polymeric Materials Polym. Chem.2023144783480310.1039/D 3PY 00997 A · doi ↗
- 6Wang J.-S. Matyjaszewski K. Controlled/”living” Radical Polymerization. Atom Transfer Radical Polymerization in the Presence of Transition-Metal Complexes J. Am. Chem. Soc.19951175614561510.1021/ja 00125 a 035 · doi ↗
- 7Kato M. Kamigaito M. Sawamoto M. Higashimura T. Polymerization of Methyl Methacrylate with the Carbon Tetrachloride/Dichlorotris-(Triphenylphosphine)Ruthenium(II)/Methylaluminum Bis(2,6-di-tert-butylphenoxide) Initiating System: Possibility of Living Radical Polymerization Macromolecules 1995281721172310.1021/ma 00109 a 056 · doi ↗
- 8Pan X. Tasdelen M.A. Laun J. Junkers T. Yagci Y. Matyjaszewski K. Photomediated Controlled Radical Polymerization Prog. Polym. Sci.2016627312510.1016/j.progpolymsci.2016.06.005 · doi ↗