PhyloJS: Bridging phylogenetics and web development with a JavaScript utility library
Leo A. Featherstone, Wytamma Wirth

TL;DR
PhyloJS is a JavaScript library for handling phylogenetic trees, enabling efficient manipulation and integration with web tools.
Contribution
PhyloJS introduces a lightweight, zero-dependency JavaScript/TypeScript library for phylogenetic tree manipulation and interoperability.
Findings
PhyloJS efficiently handles large phylogenetic trees with up to 10^6 tips.
The library is designed for interoperability with other phylogenetic and data visualization tools.
PhyloJS is available on GitHub with extensive documentation and npm installation support.
Abstract
There is an increasing number of libraries devoted to parsing, manipulating and visualising phylogenetic trees in JavaScript. Many of these libraries bundle tree manipulation with visualisation, but have limited ability to manipulate trees and lack detailed documentation. As the number of web‐based phylogenetic tools and the size of phylogenetics datasets increases, there is a need for a library that parses, writes and manipulates phylogenetic trees that is interoperable with other phylogenetic and data visualisation libraries. Here we introduce PhyloJS, a light zero‐dependency TypeScript and JavaScript library for reading, writing and manipulating phylogenetic trees. PhyloJS allows for modification of and data‐extraction from trees to integrate with other phylogenetics and data visualisation libraries. It can swiftly handle large trees, up to at least 10 6 tips in size, making it ideal…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
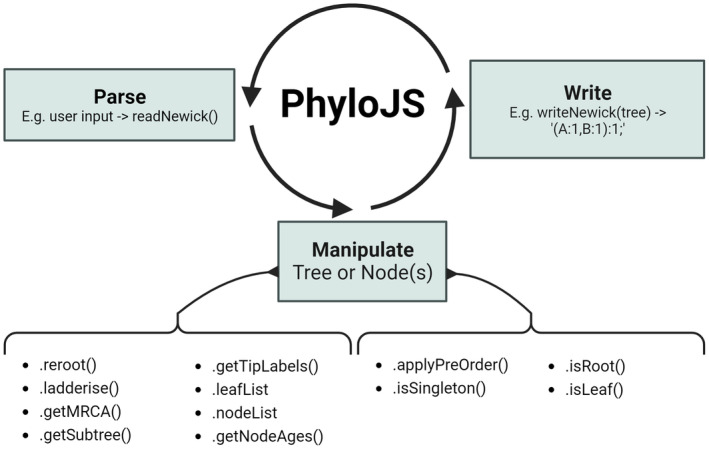 FIGURE 1
FIGURE 1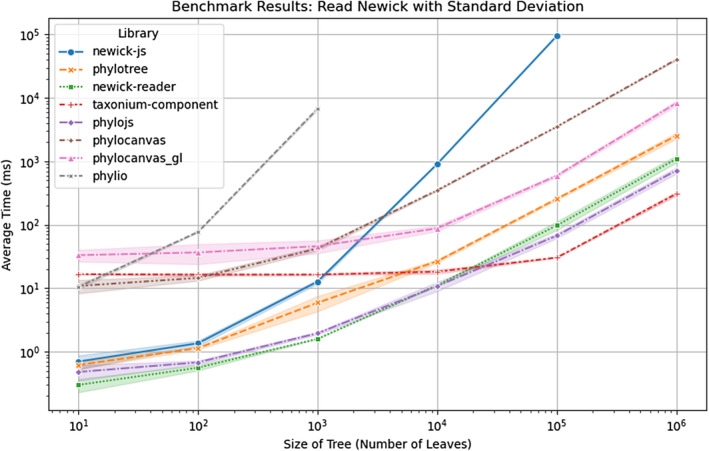 FIGURE 2
FIGURE 2| Version | Size (kb) | Dependencies | TypeScript | Networks | Newick | Nexus | phyloXML | NeXML | PhyJSON | Multiple trees | Reroot | Annotations | Extract clades | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PhyloJS | 1.5.10 | 15.1 | 0 | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| Phylotree (Shank et al., | 1.4.0 | 238.8 | 11 | N | N | Y | Y | N | N | N | N | Y | Y | N |
| Phylocanvas (Abudahab et al., | 2.8.1 | 53.6 | 0 | N | N | Y | N | N | N | N | N | Y | N | N |
| Phylio | 1.1.2 | 184 | 0 | Y | N | Y | Y | Y | N | N | N | N | Y | N |
| PhyD3 (Kreft et al., | 2.0.0 | 23.4 | 4 | Y | N | N | N | N | N | N | N | N | N | N |
| Newick‐js | 1.2.1 | 5.1 | 0 | Y | N | Y | N | N | N | N | N | N | N | N |
| newick‐reader | 1.3.0 | 1.8 | 0 | N | N | Y | N | N | N | N | N | N | N | N |
| Example | Application |
|---|---|
| Random Rerooting App | Any app requiring rerooting |
| Ladderising App | Improve visualisation for users |
| Newick Conversion App | Converting between tree formats |
| Tree statistics App | Calculate tree‐imbalance |
| Fetching trees from URLs | Accessing online trees (e.g. UShER output (Turakhia et al., |
| Root‐to‐tip Regression | Clockor2 |
| Pruning and Grafting | Removing outlier clades, topological operators in MCMC |
| Extracting Clades | Isolating clades of epidemiological interest |
| Modifying Annotations | Linking metadata |
| Annotations and Tree Traversal | Tree traversal (e.g. best fitting root in TreeTime (Sagulenko et al., |
| Arrays of Trees | Handle Posterior distributions of trees |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Plant and animal studies · Species Distribution and Climate Change
INTRODUCTION
1
Web‐based phylogenetics applications have become increasingly popular, concomitant with a general increase in web‐based bioinformatics applications. They offer notable benefit for the accessibility and scale of phylogenetic analysis. For example, Nextstrain makes the results of ongoing genomic epidemiology analyses accessible to non‐expert users through a web‐based interface (Hadfield et al., 2018). Similarly, Microreact provides a web‐based interface for the integrated visualisation of phylogenetic, spatial and epidemiological data (Argimón et al., 2016). The interactive exploration of trees with millions of tips has also been made possible with Taxonium (Sanderson, 2022). Other applications for visualising phylogenetic data include PhyD3 (Kreft et al., 2017), Phylo.io (Robinson et al., 2016) and IcyTree (Vaughan, 2017).
While many of the above applications excel at phylogenetic data visualisation and analysis, each implements its own code for parsing, writing and manipulating trees that is tightly coupled to the application itself, such that tree‐manipulating code is not separately available as a reusable package. In addition, other libraries that aim for broader utility integrate tree representation with the D3 visualisation library, inheriting dependencies and constraining their use in other applications (Bostock et al., 2011) (Table 1).
Overall, as of early September 2023 there were 57 packages matching the search term ‘phylogenetic’ in the npm registry (the main repository for JavaScript packages, akin to CRAN and PyPI for R and Python packages) from the last 10 years (Table 1). Of these, the majority became available in the last 5 years. All of these are either primarily devoted to visualisation, or offer limited ability to manipulate trees to the extent available in the state‐of‐the‐art libraries of other ecosystems such as ape or treeio in R; dendroPy or ETE3 in python; and PhyloNetworks in julia (Huerta‐Cepas et al., 2016; Paradis & Schliep, 2019; Solís‐Lemus et al., 2017; Yu et al., 2017).
Although visualisation is essential because phylogenetic trees are inherently visual models, the large number of visualisation libraries has created a niche for a library solely devoted to manipulating phylogenetic trees. Moreover, such a library further enables phylogenetic computation in client‐side applications, where all computation is done in the browser without need for a server. This has notable benefits for data security and accessibility, especially for sensitive data in genomic epidemiology since the data never leaves the user's computer. In conjunction, visualisation and manipulation‐oriented libraries will allow developers to produce applications with both high quality visualisation and greater functionality.
Here we present, PhyloJS, a well‐documented, standardised and standalone JavaScript library for parsing, writing and manipulating phylogenetic trees to develop novel phylogenetics web‐based applications with an emphasis on client‐side computation. PhyloJS focuses on tree manipulation rather than visualisation and can efficiently interface other phylogenetic and data visualisation libraries, such as phylocanvas, phylotree or plotly (Abudahab et al., 2021; Plotly‐Technologies‐Inc, 2015; Shank et al., 2018).
IMPLEMENTATION AND USAGE
2
A detailed application programming interface (API) for PhyloJS documenting all functions, methods and classes can be found at https://clockor2.github.io/phylojs/.
The representation, parsing and writing of trees in PhyloJS is based on the algorithms used in IcyTree, a well‐established client‐side visualisation tool for phylogenetic trees (Vaughan, 2017). PhyloJS is written in TypeScript, supporting type‐safe development and, therefore, is ideal for debugging larger applications. Overall, it offers a package that will be familiar to those who have used popular phylogenetic utility libraries in other languages such as ape (Paradis & Schliep, 2019) in R and DendroPy (Sukumaran & Holder, 2010) in Python (Figure 1).
Conceptual figure of PhyloJS' utility, with a non‐exhaustive list of functions and methods for parsing and manipulating trees and nodes. PhyloJS parses trees, such as from user input and other libraries, and provides a utility library to operate on the tree and return it for visualisation, user interaction and processing via other libraries.
PhyloJS includes two classes: Node and Tree (Figure 1). The Node class includes properties for branch length (branchLength) leading into the node, as well as descending and ancestral nodes (children and parent, respectively). Child and parent properties facilitate the nesting of nodes, wherein a node can have any number of descending child nodes and up to one parent (zero for the root). Node objects also store annotations via the annotation property, which is itself an object storing key‐value pairs of annotations. Hybrid nodes in phylogenetic networks are also denoted with a Boolean property (isHybrid = TRUE).
The Tree class is a wrapper of the Node class. It contains the root node at the highest level, and each subsequent node nested within it. It includes getters for internal nodes and tips (.nodeList and .leafList, respectively), which return arrays of Node objects (a leaf is a node object without any child nodes). The Tree class includes many other methods for manipulating trees, referenced in the following sections and examples.
Parsing
2.1
PhyloJS includes functions to read annotated and un‐annotated trees from all common formats (Newick, Nexus, PhyloXML, NeXML and PhyJSON), (Boettiger et al., 2016; Cardona et al., 2008; Han & Zmasek, 2009; Maddison et al., 1997; Ronquist et al., 2021). PhyloJS can also parse lists of trees separated by the newline character in formats additional to Nexus (e.g. readTreesFromNewick(), readTreesFromPhyloXML()) to arrays of Tree objects. This is useful for efficiently manipulating multiple trees at once using array methods in JavaScript (e.g. TreesArray.map(tree = > tree.ladderise())). Finally, PhyloJS includes a general function read(), that accepts strings and a schema argument to select a particular file format. It returns a Tree array (Tree[]) in all cases and is useful for applications with multiple input file formats.
Writing
2.2
PhyloJS writes individual trees to Newick and Nexus formats (writeNewick(), writeNexus()). Both functions also accept a callback for including annotations. The default behaviour is not to annotate and users can supply their own annotator or use the inbuilt beastAnnotation() or nhxAnnotation() functions to write annotations in the common formats used in BEAST or NHX (Bouckaert et al., 2019; Suchard et al., 2018). If users wish to write from an array of trees to one string, then writing functions can be applied via a JavaScript array method (e.g. TreesArray.map(tree = > writeNewick(tree)).join(’\n’)). The branch length property (.branchLength) for each node can be undefined, and all tree‐writing functions omit undefined branch lengths when writing. Last, PhyloJS does not automatically resolve polytomies and thus can write and parse multifurcating trees.
Manipulating
2.3
The Tree and Node classes include a number of methods that are unique among JavaScript libraries and or useful for manipulating trees. For topological manipulation, the Tree class includes methods for rerooting (.reroot()), ladderisation (.ladderise()) and extraction of subtrees (.getClade()). It also includes methods for accessing common ancestors (.getMRCA()).
The Node class also includes methods to add and remove children (.addChild() and .removeChild()), get ancestral nodes (.getAncestors()) and apply a function via a pre‐order and post‐order traversal of nested nodes (.applyPreOrder() and .applyPostOrder()). Branch length modifications can be applied directly to the .branchLength property of each node.
The Tree class also includes several convenience methods that help to extract data from the trees. For example, (.getRTTDist()) returns root‐to‐tip distances for each tip and (.getBranchLengths()) returns all branch lengths. For example, the root‐to‐tip regression tool Clockor2 (clockor2.github.io) uses these methods internally (Featherstone et al., 2024).
Benchmarking
2.4
We benchmarked PhyloJS against other libraries in Table 1 by parsing simulated Newick trees with 10^1^ to 10^6^ tips. Parsing Newick trees presents a task common to all phylogenetics libraries, and we note that methods on the Tree class remained fast for the largest trees. PhyloJS was among the fastest, with only a fractional delay compared to newick‐reader (<1(ms)) for trees with fewer than 10^4^ tips. This was probably due to additional logic in PhyloJS' Newick parser, which accounts for annotations and hybrid nodes and is lacking in newick‐reader (Figure 2). Taxonum‐component, though not a library for manipulating phylogenetic trees, is included as a comparison because it presents the state‐of‐the‐art parser optimised for the largest phylogenetic trees Sanderson (2022). For trees with more than 10^4^ tips, PhyloJS is second to Taxonium in speed.
A comparison of time taken in milliseconds to parse Newick trees of increasing size. Colour corresponds to each phylogenetics library and shading represents the standard deviation of time taken to parse a tree of size n across 5 duplicates. PhyloJS is among the fastest, with a minor performance loss compared to newick‐reader due to additional logic for parsing annotations and hybrid nodes. Taxonium, though not formally a library for manipulating phylogenetic trees, is included as a comparison because it is optimised for parsing large trees. Reads that took longer than 100 s to complete were cancelled.
Both PhyloJS and Taxonium incorporate a parser that utilises a stack to read Newick strings, based on the parser in jstreeview (https://github.com/lh3/jstreeview/tree/main). We found this approach conferred a notable speedup in comparison to recursive approaches and the capacity to parse much larger trees.
EXAMPLES AND DOCUMENTATION
3
Documentation for PhyloJS includes an API reference and several interactive examples at https://clockor2.github.io/phylojs/. Most examples demonstrate how to build small applications performing functions such as finding the most recent common ancestor for sets of tips, root‐to‐tip regression, rerooting, interfacing with visualisation libraries, handling multiple trees concurrently, modifying annotations, pruning and grafting clades and calculating summary statistics. Some examples also demonstrate how developers may parse trees from user input, including from URLs. Example applications also demonstrate how PhyloJS can be used to interface with visualisation libraries, such as phylocanvas (Abudahab et al., 2021) (Table 2).
Below, we include examples highlighting applications of PhyloJS, and how the code looks in practice. The final two examples demonstrate working with arrays of trees and annotating nodes with summary statistics. Together, they demonstrate PhyloJS’ ability to concurrently process trees, modify annotations, as well as modify topology and branch lengths. We urge readers wishing to run the code to view these as interactive examples at (https://clockor2.github.io/phylojs/).
Clockor2: A larger scale example
3.1
PhyloJS was initially developed to support phylogenetics web applications that incorporate analysis and visualisation without the need for server side computation. Clockor2 (https://clockor2.github.io/) is a key example, performing root‐to‐tip regression with strict local molecular clocks in the browser using PhyloJS. Here, PhyloJS provides all of the functionality to parse, manipulate and analyse trees including root‐to tip regression. All operations make use of PhyloJS, which parses and returns trees in Newick format for visualisation using phylocanvas and Plotly, or for download (Abudahab et al., 2021; Plotly‐Technologies‐Inc, 2015).
Interfacing with visualisation libraries
3.2
For a minimal example of interfacing PhyloJS with a visualisation library, we direct readers to the visualisation example https://clockor2.github.io/phylojs/examples/visualise/, which uses PhyloJS to randomly reroot a tree and phylocanvas for visualisation (Abudahab et al., 2021).
Working with arrays of trees
3.3
In the following example, we parse two trees written in Newick format to an array of Tree objects using readTreesFromNewick(). For both trees, all branch lengths are equal to 1. We arbitrarily reroot each tree at the 4th node, ladderise them and randomly rescale each branch length. We finally write the resulting trees to Newick format. This example demonstrates how users can manipulate multiple trees concurrently. // Using two small trees with 3 tips and all branch lengths set to 1. const inNewick = ’ (( A :1 , B : 1 ) : 1 , C : 1 ) ; \n (( A :1 , B : 1 ) : 1 , C : 1 ) ; ’ l e t trees = readTreesFromNewick ( inNewick ) ; // Operate on trees using array methods. Reroot , ladderise , and scale branch lengths randomly trees. forEach ( t => t. reroot ( t. nodeList [4] ) ) // ar b i t r a r i l y to 4 th node trees. forEach ( t => t. ladderise ()) trees. forEach ( t => t. nodeList. forEach ( n => n. branchLength !== undefined ? n. branchLength = Math. floor (10 Math. random () + 1) : 0 )) // write output l e t outNewick = trees. map ( t => writeNewick ( t ) ). join ( ’ \n ’ ) console. log ( outNewick ) // (" C ":1.5 ,(" B ":5 ," A ":6):4.5):0.0; // (" C ":1 ,(" B ":8 ," A ":16):4):0.0;
Internal to external branch length ratio annotation
3.4
Here, we demonstrate how to calculate the internal to external branch length ratio (IE ratio hereon) for clades descending from each internal node of a tree. We then add the statistic as a node annotation. This sort of program could, for example, be implemented as part of an application to visualise parts of a tree that drive values of summary statistics the most. // IE ratio function function getIERatio ( tree : Tree ) : number { l e t sumInternal : number = 0. 0 ; l e t sumExternal : number = 0. 0 ; for ( l e t i =0; i < tree. nodeList. length ; i ++) { if ( tree. nodeList [ i ]. branchLength !== undefined ){ if ( tree. nodeList [ i ]. isLeaf ()) { sumExternal += tree. nodeList [ i ]. branchLength } else { sumInternal += tree. nodeList [ i ]. branchLength } } } return sumInternal / sumExternal ; } l e t nwk = ‘(( a :2 , b : 2 ) : 1 , ( c :1 , d : 1 ) : 4 ) ; ‘ l e t tree = readNewick ( nwk ) // get IE ratio for subtrees descending from each node , except tips tree. root. applyPreOrder (( node : Node ) => { if ( ! node. isLeaf ()) { l e t ieRatio = getIERatio ( tree. getSubtree ( node )) node. annotation = { ieRatio : ieRatio. toFixed (2)} } }); // Expect annotations in newick with ‘ true ’ flag console. log ( writeNewick ( tree , beastAnnotation )) // Returns : // ((" a ":2 ," b ":2[&" ieRatio "="0.25"]:1(" c ":1 ," d ":1[" ieRatio "="2.00"]:4[&" ieRatio "="0.83"]:0.0;
CONCLUSIONS
4
PhyloJS helps to fill the niche for a general phylogenetics utility library in the JavaScript ecosystem. Among the diversity of libraries providing specialised functionality, particularly for visualisation, PhyloJS serves as foundation for manipulating trees and inter‐operating with other libraries to build large phylogenetics applications. It offers a mix of scale and flexibility, handling phylogenetic trees and networks from many formats up to at least 10^6^ tips in size, and including diversity of methods with which to modify and analyse trees. Together with its extensive documentation, it will help developers to build the next generation of web‐based phylogenetics applications without reinventing the wheel.
AUTHOR CONTRIBUTIONS
Leo A. Featherstone: Conceptualization (equal); software (equal); writing – original draft (lead); writing – review and editing (lead). Wytamma Wirth: Conceptualization (equal); software (lead); supervision (lead); writing – original draft (supporting); writing – review and editing (supporting).
CONFLICT OF INTEREST STATEMENT
LAF and WW declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abudahab, K. , Underwood, A. , Taylor, B. , Yeats, C. , & Aanensen, D. M. (2021). Phylocanvas.Gl: A Web GL‐powered Java Script library for large tree visualisation . Retrieved from https://osf.io/nfv 6m/
- 2Argimón, S. , Abudahab, K. , Goater, R. J. E. , Fedosejev, A. , Bhai, J. , Glasner, C. , Feil, E. J. , Holden, M. T. G. , Yeats, C. A. , Grundmann, H. , Spratt, B. G. , & Aanensen, D. M. (2016). Microreact: Visualizing and sharing data for genomic epidemiology and phylogeography. Microbial Genomics, 2(11), e 000093.28348833 10.1099/mgen.0.000093 PMC 5320705 · doi ↗ · pubmed ↗
- 3Boettiger, C. , Chamberlain, S. , Vos, R. , & Lapp, H. (2016). R Ne XML: A package for reading and writing richly annotated phylogenetic, character and trait data in r. Methods in Ecology and Evolution, 7(3), 352–357.
- 4Bostock, M. , Ogievetsky, V. , & Heer, J. (2011). D 3 Data‐Driven Documents. IEEE Transactions on Visualization and Computer Graphics, 17(12), 2301–2309. 10.1109/TVCG.2011.185 22034350 · doi ↗ · pubmed ↗
- 5Bouckaert, R. , Vaughan, T. G. , Barido‐Sottani, J. , Duchêne, S. , Fourment, M. , Gavryushkina, A. , Heled, J. , Jones, G. , Kühnert, D. , Maio, N. D. , Matschiner, M. , Mendes, F. K. , Müller, N. F. , Ogilvie, H. A. , Plessis, L. d. , Popinga, A. , Rambaut, A. , Rasmussen, D. , Siveroni, I. , … Drummond, A. J. (2019). BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. P Lo S Computational Biology, 15(4), e 1006650. 10.1371/journal.pcbi.1006650 3095881 · doi ↗ · pubmed ↗
- 6Cardona, G. , Rosselló, F. , & Valiente, G. (2008). Extended Newick: It is time for a standard representation of phylogenetic networks. BMC Bioinformatics, 9(1), 532. 10.1186/1471-2105-9-532 19077301 PMC 2621367 · doi ↗ · pubmed ↗
- 7Featherstone, L. A. , Rambaut, A. , Duchene, S. , & Wirth, W. (2024). Clockor 2: Inferring global and local strict molecular clocks using root‐to‐tip regression. Systematic Biology, syae 003. 10.1093/sysbio/syae 003 38366939 PMC 11377183 · doi ↗ · pubmed ↗
- 8Fischer, M. , Herbst, L. , Kersting, S. , Kühn, A. L. , & Wicke, K. (2023). Tree balance indices: A comprehensive survey. Springer International Publishing. 10.1007/978-3-031-39800-1 · doi ↗