A Child Plexiform Neurofibroma of the Temple Region: A Case Report
Mubarak S Alqahtani, Salmah M Alharbi, Bandar Alamri, Muayyad Alhefzi, Adel Alawwadh

TL;DR
A seven-year-old girl had a rare tumor in her temple area, which was successfully removed through surgery.
Contribution
This case report adds to the limited literature on sporadic plexiform neurofibromas in children.
Findings
The tumor began growing at birth and rapidly expanded over five years.
Surgical resection was effective for treating the symptomatic lesion.
Histopathology confirmed the diagnosis of plexiform neurofibroma.
Abstract
Plexiform neurofibroma is a rare variant of neurofibromatosis type 1. Diagnosis is challenging due to the highly variable clinical presentation. Early diagnosis is essential for appropriate treatment and prevention of complications. This report describes a sporadic solitary plexiform neurofibroma in the temporal region of a seven-year-old girl. The growth of the mass began at birth and grew steadily over five years. Subsequently, the mass began to expand rapidly. The patient underwent complete surgical resection under general anesthesia. Histopathological examination revealed a plexiform neurofibroma. In conclusion, surgical excision is the gold standard for cases with symptomatic, visible, large superficial lesions.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
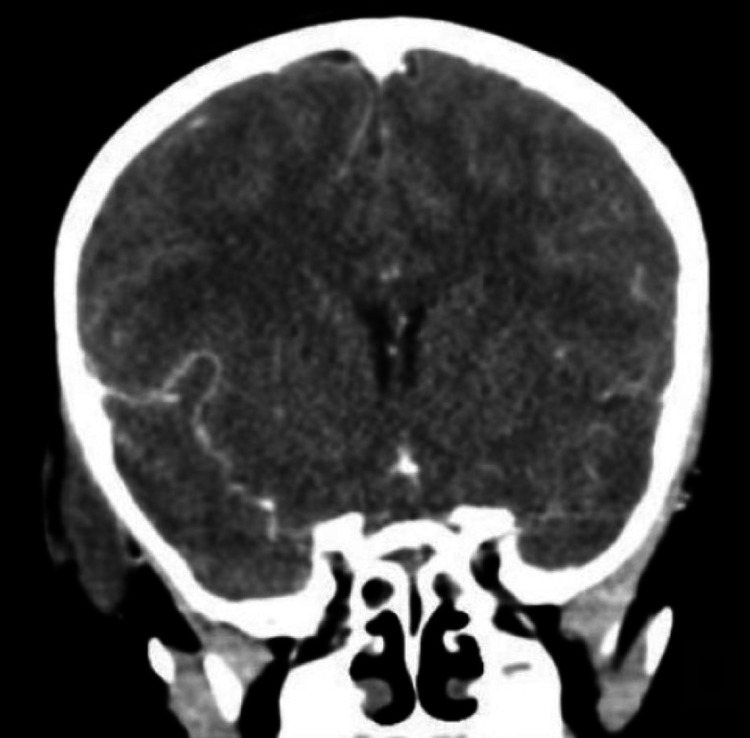 Figure 1
Figure 1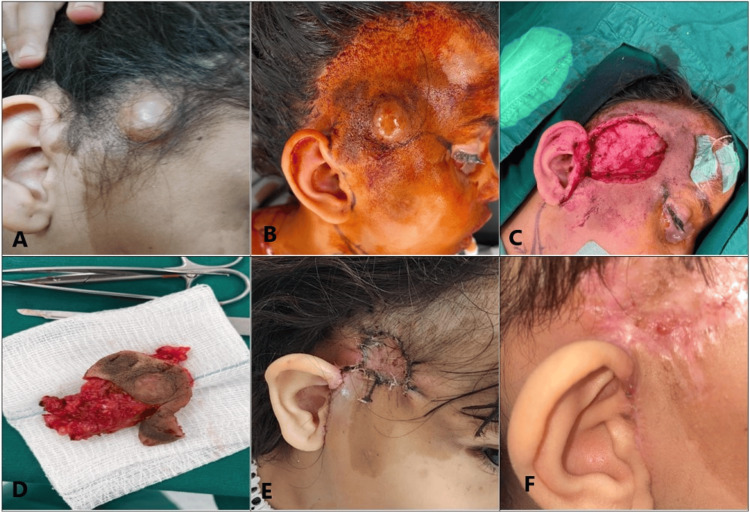 Figure 2
Figure 2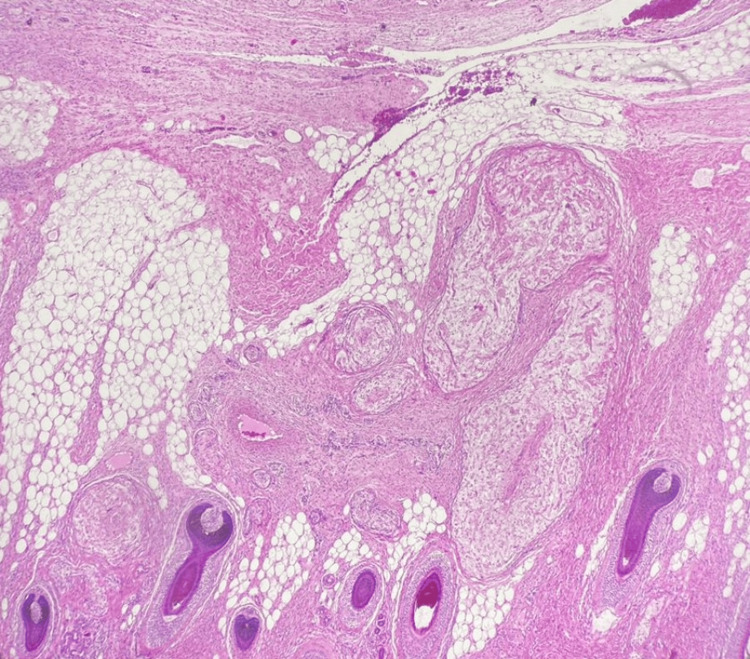 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeurofibromatosis and Schwannoma Cases · Meningioma and schwannoma management · Bone Tumor Diagnosis and Treatments