Coinheritance of non‐deletional hemoglobin H disease with sickle cell trait
Veroniki Komninaka, Evangelia‐Eleni Ntelaki

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
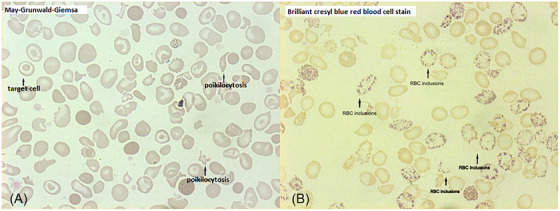 FIGURE 1
FIGURE 1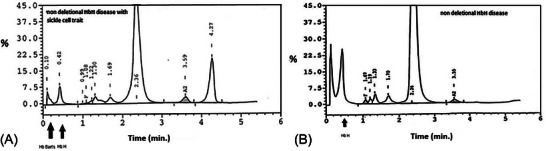 FIGURE 2
FIGURE 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHemoglobinopathies and Related Disorders · Iron Metabolism and Disorders · Erythrocyte Function and Pathophysiology
1
There are limited reports in the literature about the coinheritance of deletional hemoglobin H (HbH) disease and hemoglobin S (HbS) heterozygosity and even more limited ones about non‐deletional HbH disease and sickle cell trait [1]. We present a 51‐year‐old male of Greek origin who was referred to the clinic for anemia, splenomegaly, and fatigue investigation. The lab results were: red blood cells (RBC) 6.82 × 10^12^/L, Hb 95 g/L, hematocrit 0.35, mean corpuscular volume 55.9 fL, mean corpuscular hemoglobin 1.85 fmol, mean corpuscular hemoglobin concentration 18.5 mmol/L, red cell distribution width 0.279, RET 1.55%, white blood cells 6.2× 10^9^/L, PLT 350 × 10^9^/L, serum ferritin 0.05 nmol/L (reference values 0.04–0.4), serum Fe 13.6 nmol/L (reference values 11–22), and reduced erythrocyte life span T1/2^51^Cr 14 days. On the blood smear, we found anisocytosis, poikilocytosis, anisochromia, microcytosis, and target cells (Figure 1A). Brilliant cresyl blue RBC staining showed typical HbH inclusions (Figure 1B). The sickle cell test (with sodium metabisulphite) was positive. High‐performance liquid chromatography showed HbA 70.7%, HbS 18.5%, HbA_2_ 3.0%, HbF 0.4%, and persistence of HbH/Hb Bart's 5.2/2.8% (Figure 2A). HbA_2_ with DEAE chromatography was 1.7%. Genetic analysis using Southern blotting for the beta‐globin gene and Southern blotting for the alpha globin locus revealed HbS trait (HBB:c.20A > T) and homozygosity for a point mutation in the alpha 2 globin gene polyadenylation signal (HBA2:c.*94A > G (these findings were confirmed over time using Sanger sequencing). The radiology tests revealed splenomegaly 20 × 10 × 11 cm and osteoporosis, but no extramedullary masses, and endoscopy for gastrointestinal bleeding was negative. The patient was diagnosed with a rare combination of non‐deletional HbH disease and sickle cell trait.
(A) Blood smear morphology (B) RBC inclusions.
(A) HbH HbS HPLC (B) HbH HPLC.
It is known that α‐thalassemia with sickle cell trait results in a lower than usual percentage of HbS, a finding also detected in our patient (HbS 18.5%), who experienced no pain crisis. On the other hand, when HbH disease is inherited with sickle cell trait (β‐gene mutation) may produce hemolytic anemia with decreased or non‐detectable β‐globin tetramers HbH. The HbH percentage of our patient was 5.2% in contrast with higher HbH levels, approximately 20%–30%, in patients with HbH disease without the coinheritance of the HbS trait (Figure 2B), while in the study of Al Moamen et al. HbH ranged from 7.5% to 27.5% in the specific alpha genotype [2]. The reason is that there are decreased β^A^‐globin chains available for HbH formation since only the non‐mutant β‐globin gene synthesizes these chains. Additionally, α‐globin chains have a greater affinity for β^A^ than for β^S^‐chains, forming HbA leading to excess free β^S^‐chains. Under these terms, the lower‐than‐expected percentage of HbS in our patient is explained by the different affinity of a‐chains for β^A^ and β^S^‐chains [3, 4].
αΤSaudi homozygosity is linked to HbH clinical phenotype, despite that only two out of four α‐genes are affected. The severity of the specific mutation is proposed to be due to a transcriptional interference mechanism leading to the structurally normal α1‐globin gene downregulation [2]. The above‐mentioned lower HbH quantity could lead to a milder type of HbH disease with less ineffective hematopoiesis and therefore reduced iron overload (our patient had no iron overload) [5].
AUTHOR CONTRIBUTIONS
Conceptualization: Veroniki Komninaka; Methodology and data collection: Veroniki Komninaka and Evangelia‐Eleni Ntelaki, Supervision: Veroniki Komninaka; Writing—original draft preparation: Veroniki Komninaka; Writing—review and editing: Veroniki Komninaka. All authors have read and agreed to the published version of the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
FUNDING INFORMATION
This manuscript received no external funding.
ETHICS STATEMENT
The authors have confirmed ethical approval statement is not needed for this submission.
PATIENT CONSENT STATEMENT
All participants in this study signed consent forms.
CLINICAL TRIAL REGISTRATION
The authors have confirmed clinical trial registration is not needed for this submission.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Martinez G , Ferreira R , Hernandez A , Di Rienzo A , Colombo B . Association of Hb H disease with sickle‐trait. Hemoglobin. 1986;10(4):421–425. 10.3109/03630268608996872 3744872 · doi ↗ · pubmed ↗
- 2Al Moamen NJ , Thabet A , Mahdi F , Newton H , Salman E . Various α‐Thalassemia genotype combinations of the Saudi‐Type polyadenylation signal mutation (αT‐Saudiα) in the population of Bahrain: an update of genotype‐phenotype analyses. Hemoglobin. 2018;42(3):166–170. 10.1080/03630269.2018.1499523 30864492 · doi ↗ · pubmed ↗
- 3Matthay KK , Mentzer WC Jr , Dozy AM , Kan YW , Bainton DF . Modification of hemoglobin H disease by sickle trait. J Clin Invest. 1979;64(4):1024–1032. 10.1172/JCI 109539 479366 PMC 372212 · doi ↗ · pubmed ↗
- 4Kanavakis E , Papassotiriou I , Karagiorga M , Vrettou C , Metaxotou‐Mavrommati A , Stamoulakatou A , et al. Phenotypic and molecular diversity of haemoglobin H disease: a Greek experience. Br J Haematol. 2000;111(3):915–923.11122156 · pubmed ↗
- 5Medinger M , Saller E , Harteveld CL , Lehmann T , Graf L , Rovo A , et al. A rare case of coinheritance of Hemoglobin H disease and sickle cell trait combined with severe iron deficiency. Hematol Rep. 2011;3(3):e 30. 10.4081/hr.2011.e 30 22593821 PMC 3269802 · doi ↗ · pubmed ↗