X-linked FRMD7 gene mutation in idiopathic congenital nystagmus and its role in eye movement: A case report and literature review
Fanfei Liu, Minjin Wang, Meng Liao, Longqian Liu, Xiaoshuang Jiang

TL;DR
This paper reports a new mutation in the FRMD7 gene linked to a rare eye movement disorder and describes treatment outcomes.
Contribution
The first report of the c.686G>T mutation in the FRMD7 gene causing ICN.
Findings
A novel heterozygous missense variant (c.686G>T) was identified in the FRMD7 gene in a patient with ICN.
The mutation caused a substitution of Arg (R) with Leu (L) at position 229 (p.R229L) of the FRMD7 protein.
Treatment with occlusion therapy and surgery improved the patient's binocular vision and head posture.
Abstract
Idiopathic congenital nystagmus (ICN) is an inherited disorder characterized by uncontrollable binocular conjugating oscillation. X-linked idiopathic congenital nystagmus is one of the most prevalent types of ICN. Elucidation of the genetic mechanisms involved in ICN will enhance our understanding of its molecular etiology. We report a girl with uncontrollable binocular oscillation and anomalous head posture, then presented a novel heterozygous missense variant (c.686G>T) within the mutation-rich region of the FERM domain containing 7 (FRMD7) gene in her family member. The girl received occlusion therapy and surgical operation which balanced her binocular vision and corrected the anomalous head posture. This is the first report on a mutation (c.686G>T) caused the substitution of Arg (R) with Leu (L) at position 229 (p.R229L) of the FRMD7 protein in a patient with ICN.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
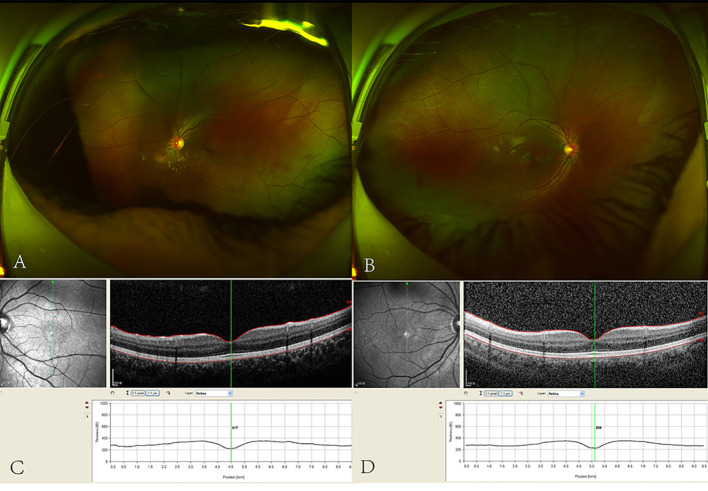 Figure 1
Figure 1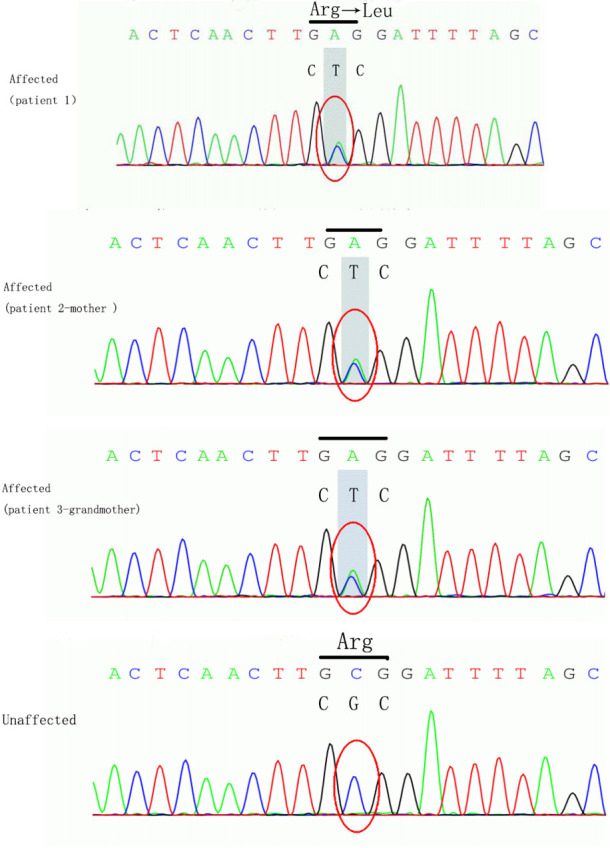 Figure 2
Figure 2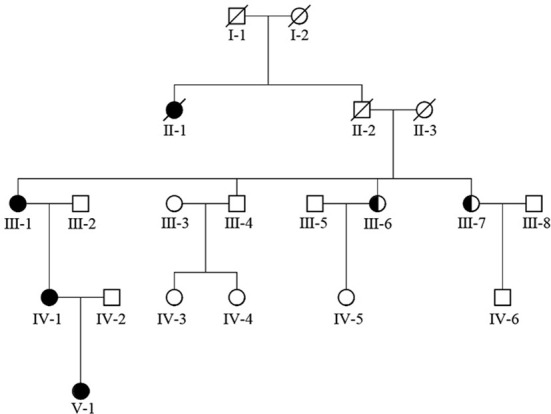 Figure 3
Figure 3| Year | Author | location | Origin | Mutation type | Nucleotide change | Protein change | References |
|---|---|---|---|---|---|---|---|
| 2020 | Fengqi Wang | Exon 12 | China | frameshift variant | c.1419_1422dup | p.Tyr475fs | ( |
| 2019 | Naihong Yan | Exon 7 | China | splice | c.498-3C > T | ( | |
| 2019 | Zhe Wang | Exon 9 | China | missense | c.805 A > C | p.Lys269Gln, K269Q | ( |
| 2018 | Yanghui Xiu | Exon 9 | China | missense | c.886G>T | p.G296C | ( |
| 2017 | Dayong Bai | Exon 11 | China | frameshift | c.999delT | p.H333fs | ( |
| 2017 | Dayong Bai | Exon 4 | China | missense | c.284G>T | p.R95M | ( |
| 2017 | Dayong Bai | Exon 4 | China | shear | c.206-1G>A | splicing | ( |
| 2017 | Dayong Bai | Exon 3 | China | shear | c.162+2T>C | splicing | ( |
| 2017 | Dayong Bai | Exon 9 | China | missense | c.782G>A | p.R261Q | ( |
| 2017 | Dayong Bai | Exon 9 | China | missense | c.811T>C | p.C271R | ( |
| 2017 | Dayong Bai | Exon 7 | China | missense | c.586G>A | p.D196N | ( |
| 2017 | Dayong Bai | Exon 10 | China | missense | c.A973G | p.R325G | ( |
| 2017 | Dayong Bai | Exon 7 | China | missense | c.521A>T | p.D174V | ( |
| 2017 | Dayong Bai | Exon 9 | China | missense | c.T766A | p.F256I | ( |
| 2017 | Dayong Bai | Exon 8 | China | missense | c.685C>T | p.R229C | ( |
| 2017 | Dayong Bai | Exon 1 | China | non frameshift | c.41_43delAGA | p.Lys14del | ( |
| 2017 | Xiuhua Jia | China | c.41_43delAGA | p.13−15delK | ( | ||
| 2017 | Xiuhua Jia | China | c.473T>A | p.I158N | ( | ||
| 2017 | Xiuhua Jia | China | c.605T>A | p.I202N | ( | ||
| 2017 | Xiuhua Jia | China | c.580G>T | p.A194S | ( | ||
| 2017 | Xiuhua Jia | China | c.811T>A | p.C271S | ( | ||
| 2017 | Xiuhua Jia | China | c.1493insA | p.Y498X | ( | ||
| 2017 | Xiuhua Jia | China | slice mutation | c.57+1G>A | ( | ||
| 2016 | Hui Zhao | Exon12 | China | nonsense | c.1090C>T | p.Q364X | ( |
| 2016 | Hui Zhao | Exon10 | China | missense | c.781C>G | p.R261G | ( |
| 2015 | Basamat AlMoallem | Exon 12 | Belgium | frameshift | c.2036del | p.(Leu679Argfs*8) | ( |
| 2015 | Basamat AlMoallem | Exon 9 | Belgium | missense | c.801C>A | p.(Phe267Leu) | ( |
| 2015 | Basamat AlMoallem | Belgium | splice-site | c.497+5G>A | ( | ||
| 2015 | Basamat AlMoallem | Exon 2 | Belgium | missense | c.70G>A | p.(Gly24Arg) | ( |
| 2015 | Basamat AlMoallem | Exon 9 | Belgium | missense | c.886G>C | p.(Gly296Arg) | ( |
| 2015 | Basamat AlMoallem | Exon 10 | Belgium | nonsense | c.910C>T | p.(Arg304*) | ( |
| 2015 | Basamat AlMoallem | Exon 8 | Belgium | frameshift | c.660del | p.(Asn221Ilefs*11) | ( |
| 2015 | Basamat AlMoallem | Exon 9 | Belgium | missense variant | c.875T>C | p.L292P | ( |
| 2015 | Shashank Gupta | Exon 7 | North Indian | missense | c.556A>G | p.M186V | ( |
| 2015 | Tomohiro Kohmoto | Exon 9 | Japan | missense variant | c.875T>C | p.L292P | ( |
| 2015 | Jae-Hwan Choi | Korean | frameshift | c.1A>G | p.Met1Val | ( | |
| 2014 | Xiao Zhang | Exon 9 | China | missense | c.780C > A | p.S260R | ( |
| 2014 | Xiao Zhang | Exon 12 | China | nonsense | c.1458 C > T | p.Q487X | ( |
| 2014 | Xiao Zhang | Exon 12 | China | deletion | c.1645del G | p.V549YfsX554 | ( |
| 2014 | Zhe Wan | Exon 9 | China | missense | c.805 A > C | p.Lys269Gln, K269Q | ( |
| 2013 | Yihua Zhu | Exon 9 | China | c.719T > C | p.I240T | ( | |
| 2013 | Zhirong Liu | Exon 6 | China | missense | c.635T>C | ( | |
| 2013 | Feng-wei Song | Exon11 | China | frameshift | c.980_983delATTA | p.H327PfsX353 | ( |
| 2013 | Feng-wei Song | Exon11 | China | frameshift | c.986C>A | p.P329E | ( |
| 2012 | Uppala Radhakrishna | Exon 10 | Switzerland | missense | c.A917G | Q305R | ( |
| 2011 | Ningdong Li | Exon7 | China | c. 623A>G | p. H208R | ( | |
| 2011 | Mervyn G. Thomas | Ireland&Gernamy | missense | c.691T>G | p.L231V | ( | |
| 2011 | Mervyn G. Thomas | Ireland | missense | c.70G>A | p.G24R | ( | |
| 2011 | Mervyn G. Thomas | England | missense | c.812G>A | p.C271Y | ( | |
| 2011 | Mervyn G. Thomas | England | missense | c.796G>C | p.A266P | ( | |
| 2011 | Mervyn G. Thomas | England | nonsense | c.1003C>T | p.R335* | ( | |
| 2011 | Mervyn G. Thomas | India | nonsense | c.1003C>T | p.R335* | ( | |
| 2011 | Mervyn G. Thomas | England | missense | c.676G>A | p.A226T | ( | |
| 2011 | Mervyn G. Thomas | England | missense | c.47T>C | p.F16S | ( | |
| 2011 | Mervyn G. Thomas | England | splice | c.58-1G>A | ( | ||
| 2011 | Mervyn G. Thomas | Romania | missense | c.1019C>T | p.S340L | ( | |
| 2011 | Mervyn G. Thomas | England | missense | c.811T>A | p.C271S | ( | |
| 2011 | Wei Du | Exon 12 | China | frameshift | c.1486–1489delTTTT | p.F497fs26X | ( |
| 2008 | Xiang He | Exon 9 | China | c.812G>T | p.C271F | ( | |
| 2008 | Ningdong Li | Exon2 | China | missense | 70 G>T | p.G24W | ( |
| 2008 | Ningdong Li | Exon8 | China | c.689–690delAG | p.Ser232del | ( | |
| 2008 | Ningdong Li | Exon9 | China | missense | c. 782G>A | p.R260Q | ( |
| 2008 | Ningdong Li | Exon9 | China | missense | c. 812G>T | p. C271F | ( |
| 2008 | Ningdong Li | Exon10 | China | nonsense | c. 910C>T | R303X | ( |
| 2008 | Xiang He | Exon 12 | China | frameshift | c.1274- 1275delTG | p.428X | ( |
| 2008 | Yuksel Kaplan | Exon 8 | Turkey | missense | c.686C>G | p.R229G | ( |
| 2007 | Qingjiong Zhang | Exon1 | China | c.41-43delAGA | p.Lys14de | ( | |
| 2007 | Qingjiong Zhang | Exon2 | China | c.70G>A | p.Gly24Arg | ( | |
| 2007 | Qingjiong Zhang | Exon6 | China | c.436C>T | p.Arg146Trp | ( | |
| 2007 | Qingjiong Zhang | Exon8 | China | c.685C>T | p.Arg229Cys | ( | |
| 2007 | Daniel F Schorderet | Exon 2 | Switzerland | nonsense | c.58C>T | p.Q20X | ( |
| 2007 | Daniel F Schorderet | Exon 9 | Switzerland | missense | c.824A>C | p.H275P | ( |
| 2007 | Daniel F Schorderet | intron 1 | Switzerland | splice | c.57+5G>A | ( | |
| 2007 | Daniel F Schorderet | intron 6 | Switzerland | splice | c.676-2A>G | ( | |
| 2007 | Daniel F Schorderet | Intron 3 | Switzerland | variant | c.206-20T>C | ( | |
| 2007 | Daniel F Schorderet | Intron 5 | Switzerland | variant | c.383-11G>A | ( | |
| 2007 | Daniel F Schorderet | Exon 8 | Switzerland | missense | c.673T>G | p.W225G | ( |
| 2007 | Alan Shiels | Exon6 | USA | missense | c.425T>G | p.L142R | ( |
| 2007 | Baorong Zhang | China | missense | c.781C>G | p.R261G | ( | |
| 2006 | Patrick S Tarpey | Ireland | missense | G70A, G24R | ( | ||
| 2006 | Patrick S Tarpey | England | truncating | IVS2+5G>A | ( | ||
| 2006 | Patrick S Tarpey | England | truncating | IVS3+2 T>G | ( | ||
| 2006 | Patrick S Tarpey | Exon4 | England | silent | G252A, V84V | ( | |
| 2006 | Patrick S Tarpey | England, England | truncating | IVS4+1G>A | ( | ||
| 2006 | Patrick S Tarpey | Ireland | missense | T425G, L142R | ( | ||
| 2006 | Patrick S Tarpey | Madagascar | truncating | IVS7+1G>C | ( | ||
| 2006 | Patrick S Tarpey | Italy-Germany | truncating | C601T, Q201X | ( | ||
| 2006 | Patrick S Tarpey | Ireland -Germany | missense | T691G, L231V | ( | ||
| 2006 | Patrick S Tarpey | England, England | missense | G796C, A266P | ( | ||
| 2006 | Patrick S Tarpey | Scotland | missense | G812A, C271Y | ( | ||
| 2006 | Patrick S Tarpey | Austria | truncating | 887delG, G296fs | ( | ||
| 2006 | Patrick S Tarpey | England | missense | A902G, Y301C | ( | ||
| 2006 | Patrick S Tarpey | England, India, England | truncating | C1003T, R335X | ( | ||
| 2006 | Patrick S Tarpey | Germany | truncating | IVS11+1G>C | ( | ||
| 2006 | Patrick S Tarpey | England | deletion | 41_43delAGA, 14delI | ( | ||
| 2006 | Patrick S Tarpey | Austria | missense | G71A, G24E | ( | ||
| 2006 | Patrick S Tarpey | England | truncating | 479insT, 160fs | ( | ||
| 2006 | Patrick S Tarpey | England | missense | A661G, N221D | ( | ||
| 2006 | Patrick S Tarpey | England | missense | G676A, A226T | ( | ||
| 2006 | Patrick S Tarpey | Romania | missense | C1019T, S340L | ( | ||
| 2006 | Patrick S Tarpey | England | truncating | 1262delC, 421fs | ( |
- —National Natural Science Foundation of China 10.13039/501100001809
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsResearch in Social Sciences
Background
Idiopathic congenital nystagmus (ICN) is a disorder characterized by uncontrollable binocular conjugating oscillation that occurs in the horizontal or vertical plane of either a jerking or pendular waveform. These pathophysiological events progress commonly at birth or shortly afterward (1). Genetically, ICN is a heterogeneous disease that could be divided into autosomal dominant (2), recessive (3, 4), and X-linked dominant (3), with X-linked type being the most prevalent (5). This disorder may be caused by deregulation of signaling pathways or mutation of a single gene (6). Two major genes, FERM domain containing 7 (FRMD7) and G-protein coupled receptor 143 (GPR143), have been implicated in the pathogenesis of ICN (7, 8). Studies have demonstrated that FRMD7 is expressed in the retina, cerebellum, lateral ventricles, and neurite during development (6). Several distinct X-linked loci have been reported to be associated with congenital nystagmus: Xp11.4-p11.3 (4), Xq26-Xq27 (3), Xp22.3-p22.2 (9), and Xq24-q26.3 (10). Approximately 20–97% of patients with X-linked ICN harbor FRMD7 mutations at Xq26.2 (11).
ICN is highly associated with the occurrence of low visual acuity, which is attributed to formation of an unstable image away from the fovea (1). Hypothetically, ICN could be a developmental response to incomplete high-contrast foveal vision (12). Most patients show a fixed angle where they achieve the best visual acuity and turn their heads in the opposite direction of the null zone. Currently, the pathogenesis of ICN has not been fully established. Although there is no etiological treatment for ICN, surgery is often performed to improve patient symptoms. We investigated a five-generation Chinese family with a history of ICN. Surgery was performed on a child diagnosed with ICN to improve anomalous head posture. A summary of recent studies exploring the association of FRMD7 gene mutation with ICN, as well as the role of FRMD7 plays in the regulation of eye movement is presented in this report.
Case presentation
This case report was approved by the ethics committee of the West China Hospital of Sichuan University and written informed consents were obtained from the patients. A four-year-old Chinese girl who presented with uncontrollable binocular oscillation and anomalous head posture was referred to the West China Hospital of Sichuan University. She had no significant past ophthalmic or medical history. On admission, her corrected visual acuity (BCVA) was 20/66 with +1.00Dc×90° in the right eye and 20/40 with +0.50 Dc×90° in the left eye. Her nystagmus was pendular and horizontal. Her face turned right to get optimal VA. No remarkable abnormalities were observed in the anterior segments of both eyes. Findings of the fundus exam and optical coherence tomography (OCT) revealed that thickness measurements of the retinal layer and macular fovea were normal (Figure 1). Moreover, titmus test showed no near stereopsis. No other general physical and neurological symptoms were observed.
Fundus photography and OCT of the four-year-old Chinese girl. (A) Fundus photography of left eye. (B) Fundus photography of right eye. (C) OCT of the macular fovea and thickness of retinal layer on left eye. (D) OCT of the macular fovea and thickness of retinal layer on right eye.
Evidence from previous studies demonstrate that refractive correction could improve visual acuity and reduce nystagmus in ICN (13).We gave the four-year-old Chinese girl a prescription for glasses and followed up. At one-year follow-up, her BCVA was 20/50 in the right eye and 20/30 in the left eye. Moreover, occlusion was observed in the left eye. At two-year follow-up, monocular BCVA was 20/30 in each eye. Binocular BCVA was 20/20 when she looked left, 20/30 when she looked forward, and 20/66 when she looked right.
Subsequently, surgery was performed on both eyes targeting the internal and external rectus muscles at the age of six. Specifically, the lateral rectus was recessed 10.0 mm from the insertion in the left eye, and the medial rectus was recessed 10.5 mm from the limbus in the right eye. One month after surgery, her compensatory head posture was improved, and the null zone was in the straight-ahead gaze; her binocular BVCA improved to 20/25 when she looked forward; the near and far binocular vision were restored. The titmus test revealed the occurrence of near gross stereopsis. Binocular vision examined using the synoptophore (Inami, Japan) showed a simultaneous perception at 0°, fusional amplitude ranging from -6° to +10°, and far stereopsis in the range of -5° to +10°. Her head posture was corrected to normal. At the age of nine years, her binocular BCVA was 20/25 when she looked forward and left. Surgery could shift the null zone of nystagmus into primary position (14), and recession or tenotomy of the four horizontal muscles could significantly improve visual function and eye movements in ICN patients (15). This study provide evidence that extraocular surgery intervention on two horizontal muscles could independently improve neurological and visual quality to achieve eye repositioning. The intervention on two horizontal muscles which also have been peformed by Dr.Muralidhar in patients with infantile nystagmus syndrome and face turn (16).
The girl in the present case came from a five-generation Chinese family, in which the mother and maternal grandmother had nystagmus, but the other family members did not develop nystagmus. Binocular BCVAs of the patient’s mother and maternal grandmother were 20/50, and both exhibited anomalous head postures. However, they did not undergo surgical treatment. None of the family members showed remarkable structural abnormalities in the eyes. To further investigate the underlying genetic cause of nystagmus, we scheduled a genomic DNA test for member of the family. Consequently, peripheral venous blood samples were collected from each family member after obtaining written informed consent. Genomic DNA was extracted from the blood samples, whole exome was isolated using the Agilent SureSelect Human All Exon Kit and then sequencing on the Agilent Hiseq sequencer. A novel missense heterozygous mutation, c.686G>T, was identified at codon 686 in exon 8 of the FRMD7 gene (Figure 2). The c.686G>T mutation caused a substitution of Arg (R) with Leu (L) at position 229 (p.R229L) of the FRMD7 protein. A gene test revealed that among the 20 family members, four females were affected and two members were carriers (Figure 3). The mutation was detected in the patient’s mother, maternal grandmother, and two sisters of the maternal grandmother (Figure 2).
The loci of mutation in FRMD7. The mutation c.686G>T caused a substitution of Arg (R) to Leu (L) at position 229 (p.R229L) of the FRMD7 protein in the girl, her mother and grand-mother.
FRMD7 mutations in this family. The family (20 members) included four affected females and two carriers.
Discussion and conclusions
Congenital nystagmus is a clinically and genetically heterogeneous disorder that affects vision (1). This disorder is characterized by idiopathic and nystagmus-related disorders. It has been postulated that ICN might be caused by a developmental defect in the brain’s ocular motor regions that control fixation (1).
Here, we identified a novel heterozygous missense mutation of the FRMD7 gene in a Chinese family with ICN. A four-year-old Chinese girl who had ICN underwent surgical treatment to reposition of the minimal intensity zone to the primary position of the eye. The surgery balanced binocular vision and corrected the anomalous head posture at 3-year follow-up. The identified mutation caused substitution of CGC (that codes for Arg) with CTC (that codes for Leu) in eight exons of position of 229 in the FRMD7 gene, c.686G>T. Two other reports of missense mutation of the position 229 have been described in FRMD7 (17, 18). One of them is a missense c.686C>G mutation in exon 8 of the FRMD7 gene, which results in the substitution of Gly(G) for Arg(R) at amino acid position 229 (p.R229G) (17). The other is a missense c.685C>T mutation, which causes the substitution of Arg (R) with Cys (C) at position 229 (p.R229C) in exon 8 of the FRMD7 gene (18). The missense mutations in position 229 of FRMD7 are mostly deleterious and attributed to the pathogenicity (18).The FRMD7 gene, located in chromosome Xq26.2, comprises 12 exons and encodes a polypeptide containing 714 residues (19). The FRMD7 protein, containing FERM-N, FERM-M, FERM-C, and FA structural domains, is a member of the 4.1 superfamily (19, 20). The identified novel mutation was located in the FERM-C domain, which has been reported to have the highest number of mutations compared with all other domains (20). This mutation was also detected in her mother, maternal grandmother, and two sisters of her maternal grandmother.
In our report family, nystagmus was inherited as an apparent X-linked dominant disorder with incomplete penetrance. Among the tested family members, there were four affected females and two obligate female carriers. Studies have been reported X-linked congenital idiopathic nystagmus pedigrees could be represented a penetrance in the range of 30–100% in female members (21, 22). The possible mechanisms for this variable penetrance include skewed X inactivation, genetic modifiers, regulation of other genes, and other factors that influence ocular motor development (22–26).
FRMD7 is mainly detected in neuronal tissue within the afferent arms of the vestibulo-ocular reflex consisting of the optic vesicle, cranial nerve VIII, vestibular ganglia, developing neural retina, and ventricular zone of the optic stalk. It is also selectively expressed in starburst amacrine cells (7, 27, 28). It is involved in the regulation of eye movement, neuronal morphogenesis, synapse function, and neurite growth (29). The FERM domain and the FERM-adjacent domain control plasma membrane and actin cytoskeleton organization suggesting that they are essential to the development of neural system and the brain region that control eye movement (30).
Several FRMD7 isoforms have been shown to play important roles during neuronal differentiation and development. The original form of FRMD7 (FRMD7-FL) and its splice variants FRMD7-S and FRMD7_SV2 are involved in the neuronal development process (29) (31). A search performed on the BLAST tool showed that the FRMD7 protein shared a close homology with FARP1 and FARP2, which modulate the length and the degree of branching of neurites in rat embryonic cortical neurons and reorganize the cytoskeleton (32, 33).
Evidence shows that FRMD7 mutations are associated with the development of nystagmus (34). Besides, FRMD7 mutations could alter retinal direction selectivity, diminish responsiveness at higher stimulus speeds, and eliminate overrepresentation of posterior-motion-preferring cortical cells (35), thereby cause retinal neuron migratory defects such as foveal hypoplasia (36). In addition, they could affect starburst amacrine cells and impair the development of visual motion responses in superior colliculus neurons downstream of the retina (37). This disrupts axogenesis, dendritogenesis, and neuronal guidance in brain areas involved in the control of eye movement (30). FRMD7 promoted neurite elongation by modulating actin cytoskeleton whereas FRMD7 gene knockdown led to a significant reduction in overall neurite length (34, 38), further contributing to the development of neuronal circuit asymmetry and neurological disorders (28, 39). Mutations associated with ICN include missense mutations, null mutations, deletions and insertions, and frameshift mutations. A series of FRMD7 gene mutations linked to ICN are summarized in Table 1.
In conclusion, our study adds to the spectrum of FRMD7 mutations associated with X-linked ICN. We show that FRMD7 mutations underlie the development of X-linked ICN. Surgery could be an effective treatment approach to correct persistent anomalous head posture in ICN patients. However, the molecular mechanisms by which genetic variations cause ICN are unknown, and therefore, further investigations are required to reveal detailed mechanisms that drive the pathogenesis of this hereditary ocular disease.
This study has several potential limitations. First, only one Chinese family was investigated in the study. In addition, we did not examine protein and cell levels to clarify the pathological mechanism by which FRMD7 mutation led to ICN. Furthermore, the follow-up duration was short. Therefore, further studies with longer follow-up duration are needed to validate the efficacy of surgical therapy in ICN patients.
Data availability statement
The original contributions presented in the study are included in the article/supplementary materials. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by West China Hospital of Sichuan University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
XJ and FL carried out the experiments, prepared the figures, and drafted the manuscript. MW performed the gene test. ML and LL participated in its design and edited the manuscript. All authors contributed to the article and approved the submitted version.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Papageorgiou E Mc Lean RJ Gottlob I. Nystagmus in childhood. Pediatr Neonatol (2014) 55(5):341–51. doi: 10.1016/j.pedneo.2014.02.007 25086850 · doi ↗ · pubmed ↗
- 2Kerrison JB Arnould VJ Barmada MM Koenekoop RK Schmeckpeper BJ Maumenee IH. A gene for autosomal dominant congenital nystagmus localizes to 6p 12. Genomics (1996) 33(3):523–6. doi: 10.1006/geno.1996.0229 8661013 · doi ↗ · pubmed ↗
- 3Kerrison JB Vagefi MR Barmada MM Maumenee IH. Congenital motor nystagmus linked to Xq 26-q 27. Am J Hum Genet (1999) 64(2):600–7. doi: 10.1086/302244 PMC 13777719973299 · doi ↗ · pubmed ↗
- 4Cabot A Rozet J-M Gerber S Perrault I Ducroq D Smahi A. A gene for X-linked idiopathic congenital nystagmus (NYS 1) maps to chromosome Xp 11. 4-p 11. 3. Am J Hum Genet (1999) 64(4):1141–6. doi: 10.1086/302324 PMC 137783810090899 · doi ↗ · pubmed ↗
- 5Oetting WS Armstrong CM Holleschau AM De Wan AT Summers CG. Evidence for genetic heterogeneity in families with congenital motor nystagmus (CN). Ophthalmic Genet (2000) 21(4):227–33. doi: 10.1076/1381-6810(200012)2141-HFT 227 11135493 · doi ↗ · pubmed ↗
- 6Kumar A Gottlob I Mclean RJ Thomas S Thomas MG Proudlock FA. Clinical and oculomotor characteristics of albinism compared to FRMD 7 associated infantile nystagmus. Invest Ophthalmol Visual Sci (2011) 52(5):2306–13. doi: 10.1167/iovs.10-5685 21220551 · doi ↗ · pubmed ↗
- 7Tarpey P Thomas S Sarvananthan N Mallya U Lisgo S Talbot CJ. Mutations in FRMD 7, a newly identified member of the FERM family, cause X-linked idiopathic congenital nystagmus. Nat Genet (2006) 38(11):1242–4. doi: 10.1038/ng 1893 PMC 259260017013395 · doi ↗ · pubmed ↗
- 8Hu J Liang D Xue J Liu J Wu L. A novel GPR 143 splicing mutation in a Chinese family with X-linked congenital nystagmus. Mol Vision (2011) 17:715.PMC 306015621423867 · pubmed ↗