Troponin T Assessment Allows for Identification of Mutation Carriers among Young Relatives of Patients with LMNA-Related Dilated Cardiomyopathy
Przemysław Chmielewski, Ilona Kowalik, Grażyna Truszkowska, Ewa Michalak, Joanna Ponińska, Agnieszka Sadowska, Katarzyna Kalin, Krzysztof Jaworski, Ilona Minota, Jolanta Krzysztoń-Russjan, Tomasz Zieliński, Rafał Płoski, Zofia Teresa Bilińska

TL;DR
This study shows that measuring high-sensitivity cardiac troponin T (hscTnT) can help identify young relatives of patients with LMNA-related dilated cardiomyopathy who carry the mutation.
Contribution
The study introduces hscTnT as a reliable, accessible biomarker for identifying LMNA mutation carriers in asymptomatic relatives.
Findings
High-sensitivity cardiac troponin T (hscTnT) levels were significantly higher in LMNA mutation carriers compared to controls.
An hscTnT cut-off of 5.5 ng/L achieved 86% sensitivity and 93% specificity in identifying mutation carriers.
Abstract
Background: LMNA-related dilated cardiomyopathy (LMNA-DCM) caused by mutations in the lamin A/C gene (LMNA) is one of the most common forms of hereditary DCM. Due to the high risk of mutation transmission to offspring and the high incidence of ventricular arrhythmia and sudden death even before the onset of heart failure symptoms, it is very important to identify LMNA-mutation carriers. However, many relatives of LMNA-DCM patients do not report to specialized centers for clinical or genetic screening. Therefore, an easily available tool to identify at-risk subjects is needed. Methods: We compared two cohorts of young, asymptomatic relatives of DCM patients who reported for screening: 29 LMNA mutation carriers and 43 individuals from the control group. Receiver operating characteristic (ROC) curves for potential indicators of mutation carriership status were analyzed. Results: PR…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
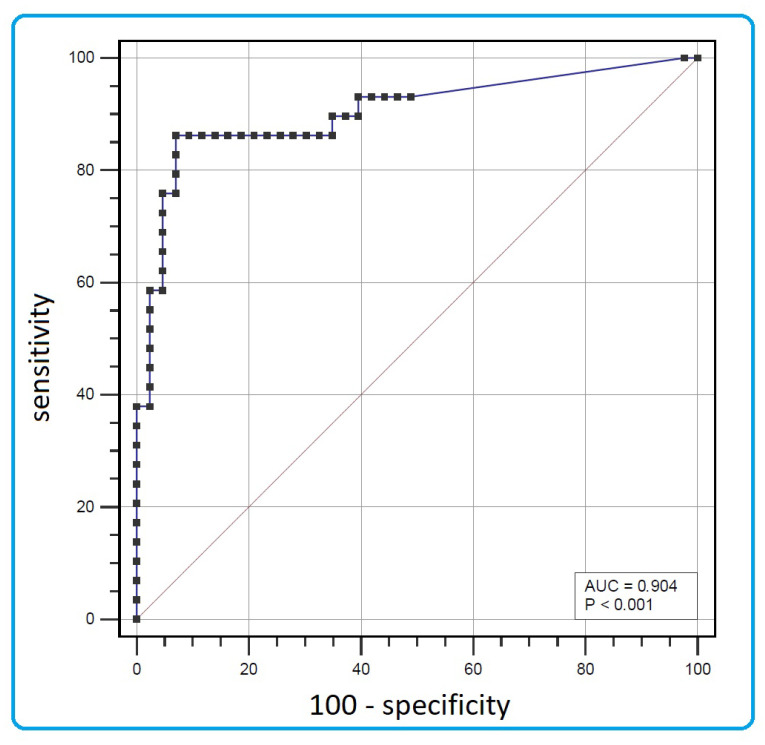 Figure 1
Figure 1- —National Institute of Cardiology, Warsaw, Poland
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLeaf Properties and Growth Measurement · Plant Physiology and Cultivation Studies · Potato Plant Research
1. Introduction
LMNA-related dilated cardiomyopathy (LMNA-DCM), caused by pathogenic variants in the lamin A/C gene (LMNA), is one of the most common forms of hereditary DCM, accounting for approximately 6% of DCM cases [1,2,3]. It is inherited in an autosomal dominant manner, so the risk of variant transmission to offspring is high (50%). LMNA-DCM is characterized by early onset, a frequent occurrence of cardiac conduction defects and arrhythmias, and an extremely unfavorable prognosis [4,5,6,7]. Therefore, relatives of LMNA-DCM patients should be subjected to clinical and genetic screening. Carriers of causative LMNA variants should be advised about lifestyle modifications, including career counselling and limiting physical activity, and should undergo periodic diagnostic tests, including standard ECG, echo, and ECG Holter recording [2,8]. Due to the high risk of ventricular arrhythmia and sudden cardiac death even before the onset of heart failure, it is very important to identify asymptomatic LMNA-mutation carriers. Some of them, despite normal left ventricular systolic function, may require protection with an implantable cardioverter-defibrillator. An arrhythmic risk calculator dedicated to LMNA-related cardiomyopathy is available [9].
Despite this, many relatives do not report for clinical or genetic screening. In many cases, psychological factors can be decisive. In other cases, access to specialist medical care may be hindered, and in many places, genetic testing, in particular, is still poorly accessible.
Therefore, an easily available tool to identify subjects at risk could be very useful. In a study published in 2020, we showed that elevated serum levels of cardiac troponin T are often found in young LMNA mutation carriers as the first abnormality, preceding conduction defects and arrhythmias [10]. In this study, we aimed to investigate whether the assessment of cardiac troponin T serum level using a high-sensitivity assay (hscTnT) helps to identify mutation carriers among young (<45 years of age), asymptomatic relatives of LMNA-DCM patients.
2. Materials and Methods
This study was conducted retrospectively and prospectively and included young (aged 18–45), asymptomatic or scantly symptomatic relatives of patients with the two most common forms of inherited DCM, LMNA-DCM and DCM associated with truncating variants of the titin gene (TTN), who reported to our unit for screening between 2013 and 2024. Previously known heart diseases, e.g., arrhythmia, as well as diseases that could affect the results of the tests, e.g., chronic kidney disease, were exclusion criteria. Non-specific and mild symptoms, such as heart palpitations or subjectively unsatisfactory physical performance, were acceptable if they did not constitute a reason to seek medical advice. All LMNA and TTN variants identified in the probands were pathogenic or likely pathogenic according to the American College of Medical Genetics and Genomics (ACMG) criteria (Table 1) [11]. All the participants underwent an interview, physical examination, 12-lead electrocardiography (ECG), echocardiography, and blood collection for genetic and biochemical testing. Genetic testing only included Sanger sequencing for the variant identified in the proband.
The study group consisted of asymptomatic carriers of a causative LMNA variant, and the control group of relatives in whom the presence of a causative LMNA or TTN variant was excluded. We compared selected parameters that could be potential indicators of LMNA mutation carrier status between the study and control groups. These included serum measurements of creatine kinase activity (CK), concentration of N-terminal prohormone B-type natriuretic peptide (NT-proBNP), and high-sensitivity cardiac troponin T (hscTnT), as well as PR interval measured on a standard ECG, and left-ventricular ejection fraction (LVEF) assessed with biplane Simpson’s method. The serum levels of NT-proBNP and hscTnT were measured by using the electrochemiluminescence immunoassays Elecsys proBNP II and Elecsys Troponin T hs STAT (both Roche, Mannheim, Germany), respectively.
All the results for the categorical variables were presented as numbers and percentages and, for continuous variables, as mean and standard deviation (SD) or median and quartiles (Q1:25th–Q2:75th percentiles). Fisher’s exact test was used for the comparison of categorical variables. The differences between continuous variables were tested by using the independent Student’s t-test (for two independent samples and for paired observation, appropriate, normally distributed data) or, in the case of skewed distribution, non-parametric Mann–Whitney U tests.
A receiver operating characteristic curve (ROC) analysis was used to assess the cut-off point for the prediction of a variant carriership. The optimal cut-off was defined as the value with the maximal sum of sensitivity and specificity (Youden’s index).
All statistical analyses were performed using SAS 9.4 (Durham, NC, USA).
3. Results
The study group consisted of 29 asymptomatic LMNA mutation carriers from 15 families, and the control group was composed of 43 mutation-free relatives from 25 families of patients with LMNA and TTN-related DCM (9 and 16 families, respectively). LMNA mutation carriers were younger, and both groups did not differ significantly concerning LVEF (Table 2). The hscTnt level was below the detection threshold (<3.0 ng/L) in most subjects from the control group. In the LMNA mutation carriers’ group, it was usually still within the normal range but significantly higher (median 11 ng/L, p <0.001; normal range <14 ng/L). PR interval and NT-proBNP serum levels were also higher in the LMNA mutation carriers’ cohort.
Based on the C-statistics and Youden’s indices, the best LMNA mutation carriership indicator was the hscTnT level (Table 3).
The optimal hscTnT cut-off value for LMNA mutation detection was 5.5 ng/L, with a sensitivity of 86% and specificity of 93% (Figure 1). Other potential discriminators were characterized by low sensitivity (50–59%) at the optimal cut-off points.
Of note, these findings were specific to LMNA mutation carriers. Asymptomatic carriers of DCM-causative TTN variants did not differ from the control group in terms of NT-proBNP and hscTnT levels (29 vs. 19 pg/mL, p = 0.20 and <3.0 vs. <3.0 ng/L, p = 0.81, respectively), despite significantly lower LVEF (Table 4).
4. Discussion
The principal new finding of our study is that the measurement of hscTnT serum concentration can be reliably used to determine young relatives of LMNA-DCM patients that may be carriers of the causative LMNA variant and that should therefore be subject to medical surveillance. We are far from saying that it can replace genetic testing; however, a hscTnT level > 5.5 ng/L has an excellent sensitivity and specificity of 86% and 93%, respectively, in detecting LMNA variant carrier status.
This finding may be treated as a “red flag” for several reasons. First, it can be used, whenever access to genetic testing is difficult, to emphasize the need for periodic screening. On the other hand, it could expedite genetic testing wherever it is feasible. Furthermore, hscTnT measurement may also be used when relatives do not report for screening due to procedural reasons (need to obtain referrals, waiting time, need to make several visits). The great advantage of troponin measurement is its widespread availability and low cost.
A prolonged PR interval as well as increased CK activity, reflecting skeletal muscle involvement, are often considered a marker of LMNA-DCM. Based on our analysis, hscTnT is a better discriminator of asymptomatic LMNA variant carrier status than those markers.
It may seem arguable that the hscTnT concentration values found in asymptomatic LMNA mutation carriers are usually still within normal limits. However, it should be noted that hscTnT levels are usually very low and often undetectable in young people. In a multicenter study, the mean hscTnT concentration was 4.0 ng/L in a cohort of 533 healthy individuals aged 20–71 [12]. The mean hscTnT levels were 2.7 and 2.6 ng/L in the case of healthy women aged 20–29 and 30–39, respectively, and 4.5 and 4.3 ng/L in the case of healthy men aged 20–29 and 30–39, respectively [12]. In the case of our study, the small study group did not allow us to conduct separate analyses for both genders.
The hscTnT values observed in the asymptomatic LMNA mutation carriers, although usually still normal, are substantially higher. This observation is consistent with the results of our previous study, in which we showed that elevated hscTnT serum levels (>14 ng/L) are often found in young LMNA mutation carriers as the first abnormality, preceding conduction defects and arrhythmias [8]. Nonetheless, this observation cannot be extrapolated to other forms of hereditary DCM. In our study on TTN-related DCM, elevated hscTnT levels could only be detected later, in the end-stage phase of the disease [13]. In asymptomatic TTN mutation carriers, we did not detect higher hscTnT levels compared to the control group during routine screening.
Cardiac troponin release can occur in many clinical situations, which may make the interpretation of test results more difficult [14,15]. It primarily occurs in many acute conditions; however, these are extremely unlikely in the context of screening a young and asymptomatic population. Other potential causes include chronic and often scantly symptomatic diseases, such as hypertrophic cardiomyopathy, valvular defects, or chronic renal failure, which should be considered during subsequent diagnostic workup [14,15,16]. Additionally, hscTnT levels may also increase after strenuous exercise [17].
False-positive troponin elevation should also be considered. It may result from the presence of heterophilic or human anti-animal antibodies, which may be present due to previous therapy with monoclonal antibodies, vaccinations, or even pet keeping. It has also been reported that false-positive results may be a consequence of the competitive interaction of diagnostic antibodies with fibrin clots, autoantibodies, or skeletal troponin molecules [15,18]. Nevertheless, in this study’s cohort, the LMNA variant carrier status was by far the most likely explanation for the higher hscTnT levels.
In LMNA-related DCM, an elevated hscTnT concentration could reflect subtle ongoing cardiomyocyte injury; however, the precise mechanisms of troponin leakage are poorly understood. It is believed that the nucleus acts as a mechanosensor through its connection to the cytoskeleton and extracellular matrix [19]. Abnormal lamins could lead to a substantial disruption of nuclear mechanobiological processes [19,20,21] and, in some situations, to obliteration of nuclear architecture [22]. The aftermath of prolonged cardiomyocyte injury is non-specific replacement fibrosis, as seen in endomyocardial biopsy or as late gadolinium enhancement in the CMR study [23,24,25]. Consequently, myocardial fibrosis is considered to be responsible for the development of electrical instability, leading to arrhythmia [6,26,27,28,29,30,31], and, over time, to mechanical impairment.
It would be worth investigating whether cardiac troponin I levels are also higher in LMNA mutation carriers. It should be noted that unlike hscTnT, which is measured by only one commercially available test, many different cardiac troponin I tests are available, including point-of-care devices.
It seems that the potential of serum biomarkers in the evaluation of patients with cardiomyopathies is not fully utilized. They are routinely used in diagnostics, and to a lesser extent in risk stratification [16,32,33]. Our study shows that their assessment may also provide additional information in the context of particular forms of hereditary cardiomyopathies [34].
5. Conclusions
Whenever access to genetic testing is limited, LMNA mutation carrier status can be reliably assessed using the hscTnT assay. Among young asymptomatic relatives of LMNA-DCM patients, a hscTnT level >5.5 ng/L strongly suggests mutation carriers. Relatives with higher hscTnT levels should be prioritized for genetic testing.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Elliott P. Andersson B. Arbustini E. Bilinska Z. Cecchi F. Charron P. Dubourg O. Kühl U. Maisch B. Mc Kenna W.J. Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases Eur. Heart J.20082927027610.1093/eurheartj/ehm 34217916581 · doi ↗ · pubmed ↗
- 2Arbelo E. Protonotarios A. Gimeno J.R. Arbustini E. Barriales-Villa R. Basso C. Bezzina C.R. Biagini E. Blom N.A. de Boer R.A. 2023 ESC Guidelines for the management of cardiomyopathies Eur. Heart J.202344350336263762265710.1093/eurheartj/ehad 194 · doi ↗ · pubmed ↗
- 3Hershberger R.E. Cowan J. Jordan E. Kinnamon D.D. The Complex and Diverse Genetic Architecture of Dilated Cardiomyopathy Circ. Res.20211281514153210.1161/CIRCRESAHA.121.31815733983834 PMC 8158434 · doi ↗ · pubmed ↗
- 4Taylor M.R. Fain P.R. Sinagra G. Robinson M.L. Robertson A.D. Carniel E. Di Lenarda A. Bohlmeyer T.J. Ferguson D.A. Brodsky G.L. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations J. Am. Coll. Cardiol.20034177178010.1016/S 0735-1097(02)02954-612628721 · doi ↗ · pubmed ↗
- 5van Rijsingen I.A. Arbustini E. Elliott P.M. Mogensen J. Hermans-van Ast J.F. van der Kooi A.J. van Tintelen J.P. van den Berg M.P. Pilotto A. Pasotti M. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study J. Am. Coll. Cardiol.20125949350010.1016/j.jacc.2011.08.07822281253 · doi ↗ · pubmed ↗
- 6Kumar S. Baldinger S.H. Gandjbakhch E. Maury P. Sellal J.M. Androulakis A.F. Waintraub X. Charron P. Rollin A. Richard P. Long-Term Arrhythmic and Nonarrhythmic Outcomes of Lamin A/C Mutation Carriers J. Am. Coll. Cardiol.2016682299230710.1016/j.jacc.2016.08.05827884249 · doi ↗ · pubmed ↗
- 7Mariani M.V. Pierucci N. Fanisio F. Laviola D. Silvetti G. Piro A. La Fazia V.M. Chimenti C. Rebecchi M. Drago F. Inherited Arrhythmias in the Pediatric Population: An Updated Overview Medicina 2024609410.3390/medicina 6001009438256355 PMC 10819657 · doi ↗ · pubmed ↗
- 8Skjølsvik E.T. Hasselberg N.E. Dejgaard L.A. LieØ.H. Andersen K. Holm T. Edvardsen T. Haugaa K.H. Exercise is Associated With Impaired Left Ventricular Systolic Function in Patients with Lamin A/C Genotype J. Am. Heart Assoc.20209 e 01293710.1161/JAHA.119.01293731957533 PMC 7033829 · doi ↗ · pubmed ↗