Late-Onset Leigh Syndrome With Protracted Gastrointestinal Manifestations: A Rare Case Report
Zain Amar, Helai Hussaini, Meet Popatbhai Kachhadia, Iqra Samreen, Alaa S Mohamed, Hira Nasir

TL;DR
A 42-year-old woman with late-onset Leigh syndrome showed unusual gastrointestinal symptoms and responded well to thiamine treatment.
Contribution
This case report highlights a rare instance of adult-onset Leigh syndrome with gastrointestinal symptoms and successful thiamine treatment.
Findings
The patient showed hyperintensities in the basal ganglia and brain stem on MRI.
Elevated lactate levels in serum and cerebrospinal fluid were observed.
Muscle biopsy revealed reduced cytochrome oxidase activity, supporting the LS diagnosis.
Abstract
Although Leigh syndrome (LS) is a neurodegenerative disorder of infancy, adult-onset LS has also been rarely reported. We report a case of late-onset LS in a 42-year-old female who presented with protracted gastrointestinal manifestations, chronic headaches, ataxia, and loss of consciousness. Brain magnetic resonance imaging (MRI) revealed hyperintensities in the bilateral basal ganglia and brain stem. Serum and cerebrospinal fluid lactate levels were significantly raised. Muscle biopsy showed reduced cytochrome oxidase (COX) activity. She was diagnosed with probable diagnosis of late-onset LS based on her clinical features, radiological findings, biochemical results, and biopsy findings. She responded well to intravenous thiamine, and her symptoms gradually improved.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
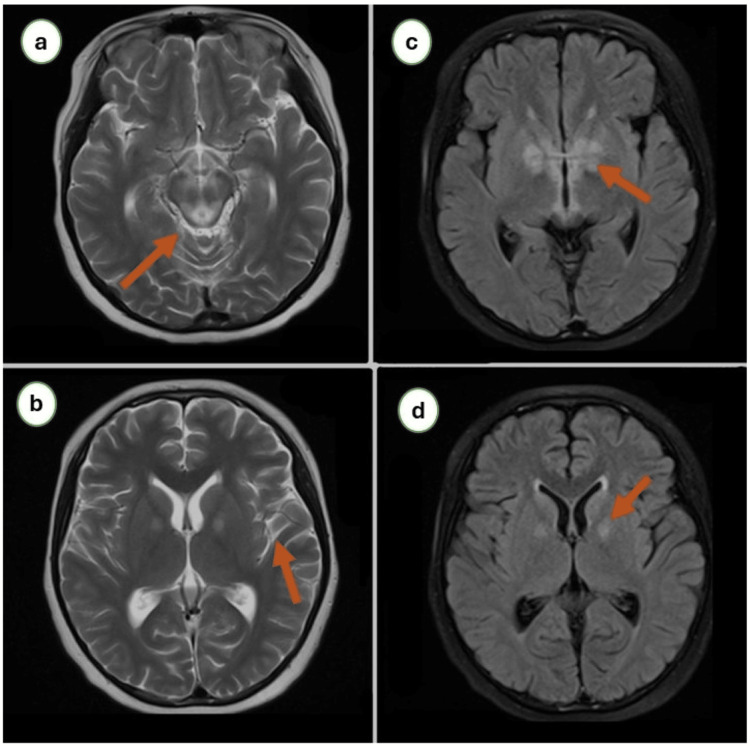 Figure 1
Figure 1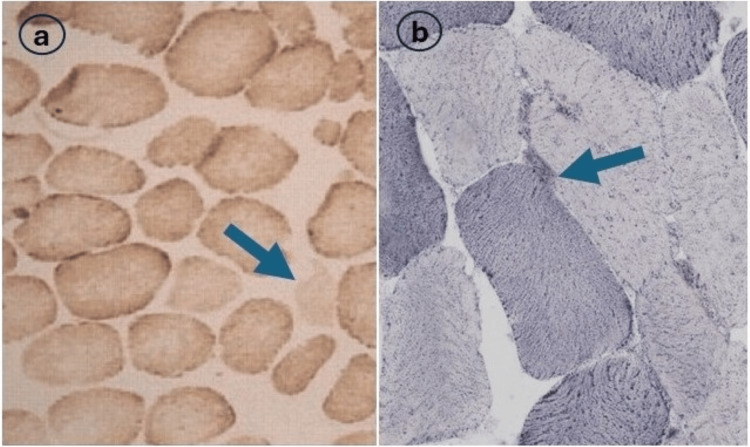 Figure 2
Figure 2| Parameter | Lab value | Reference value |
| Hemoglobin | 12.5 | 13-15 g/dl |
| White cell count | 8700 | 4000-11000/mm3 |
| Red cell count | 4.3 | 4.2-5.2 million cells/mcl |
| Alanine aminotransferase | 42 | 10-55 IU/L |
| Aspartate aminotransferase | 36 | 8-39 IU/l |
| Serum creatinine | 0.9 | 0.7-1.3 mg/dl |
| Platelet count | 210,000 | 150,000-350,000/mcl |
| Blood urea nitrogen | 22 | 7-28 mg/dl |
| Calcium | 8.9 | 8.5-10.5 mg/dl |
| C-reactive protein | 1.1 | 0.4-1.2 mg/dl |
| Serum lactate | 8.1 | 0.2-2.4 mmol/l |
| Sodium | 138 | 138-145 mEq/l |
| Potassium | 4.1 | 3.5-4.5 mEq/l |
| pH | 7.12 | 7.35-7.45 |
| Author et al. | Age/sex | Family history | Birth history | Presenting symptom | Organ involvement | Serum lactate level (mmol/L) | Muscle biopsy obtained | Mitochondrial gene mutation | MRI findings | Outcome |
| Goldenberg et al. [ | 24/F | No | Normal | Headache | CNS, respiratory | 8 | Yes, COX-negative | Yes | Brain stem hyperintensity | Regression |
| Cipriano et al. [ | 36/M | No | Normal | Walking difficulty | CNS, respiratory | 7.8 | Yes, COX-negative | No | Brain stem, basal ganglia hyperintensity | Regression |
| Wesolowska et al. [ | 45/M | No | Normal | History of fall | CNS, respiratory | 3.7 | Yes, reduced COX activity | No | Brain stem, basal ganglia hyperintensity | Ventilatory support |
| Lekha et al. [ | 26/M | No | Normal | Chronic headache | CNS | 6.8 | Yes, reduced COX activity | No | Midbrain, basal ganglia, grey matter hyperintensity | Regression |
| Liang et al. [ | 12/M | No | Normal | Blepharoptosis | CNS | 4.4 | NR | No | White matter, basal ganglia, midbrain hyperintensity | Died |
| Malogcic et al. [ | 21/F | Down syndrome | Normal | Headache, vomiting | CNS | 11.9 | Subsarcolemmal aggregations | Yes | Basal ganglia hyperintensity | Regression |
| Li et al. [ | 24/F | No | Normal | Seizure | CNS | NR | Subsarclolemmal aggregations | Yes | Basal ganglia, white matter hyperintensity | Died |
| Parameter | Findings |
| Clinical history and physical examination | Failure to thrive, mental retardation, pyramidal signs, ophthalmoplegia, dysarthria, deafness, or other neurological manifestations |
| Imaging (CT, MRI) | Lesions in bilateral basal ganglia, or brain stem, |
| Biochemical parameter | Elevated serum or CSF lactate level |
| Muscle biopsy | Mitochondrial abnormalities, mitochondrial gene mutations |
| Rule out | Multiple sclerosis, infections, metabolic disorders, toxins, and Wernicke’s encephalopathy |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMitochondrial Function and Pathology · Metabolism and Genetic Disorders · Alcoholism and Thiamine Deficiency
Introduction
Leigh syndrome (LS) is an uncommon neurogenerative disorder of infancy, characterized by subacute necrotizing encephalopathy and bilateral symmetrical gray matter necrotizing lesions in the basal ganglia, brain stem, and cerebellum [1]. LS typically presents in infancy, with over 50% of cases in the first year of life, mainly before six months of age, with a low prevalence of 1:40,000 births [2]. LS is a mitochondrial disorder that may be sporadic or familial, characterized by psychomotor regression or delay with lesions mainly in the brain stem and basal ganglia [3]. Adult-onset LS, also called late-onset LS, has been underlined in the literature, with only a few cases published. In adult-onset LS, symptoms may appear after two years and not manifest until early adulthood. Compared to the infantile form, adult-onset LS progresses slowly and is more prevalent in males [4,5]. Herein, we report a case of adult-onset LS in a female who presented with protracted gastrointestinal manifestations.
Case presentation
A 43-year-old female presented with protracted generalized abdominal pain and intermittent vomiting for the last two months, for which she used over-the-counter medications with no improvements. She also complained of multiple episodes of throbbing headaches in the last year and was treated as migraine. Now, she presented with a worsening headache for the last week, which was generalized with no aggravating and relieving factors associated with bilateral ptosis and lightheadedness. She reported no history of febrile illness, seizures, limb weakness, sensory disturbance, or other preceding illnesses. She had no personal or family history of any similar disease. She reported no history of smoking or substance abuse but sporadic alcohol use.
She was hemodynamically stable and well-oriented to time, place, and person on examination. Eye examination revealed bilateral ptosis with horizontal gaze palsy and normal pupil. Fundus examination was unremarkable, with no signs of meningeal irritation. Cranial nerve examination was normal, and neurological examination showed ataxia. The rest of the systemic examination was unremarkable.
She underwent brain magnetic resonance imaging (MRI), revealing hyperintense signals in a widespread brain area. Electroencephalography (EEG) showed generalized slowing with no epileptiform activity. Owing to her protracted vomiting and MRI findings, she was diagnosed with a probable diagnosis of Wernicke’s encephalopathy. She was managed with parenteral thiamine, showed marked improvements in her symptomology, and was discharged.
Two weeks later, she presented again with a severe headache followed by a loss of consciousness. On examination, she had brisk tendon reflexes and extensor plantar reflexes bilaterally. Her lab results were normal except for elevated fasting serum lactate level (Table 1). Arterial blood gas analysis revealed severe metabolic acidosis with a pH of 7.12. A repeat brain MRI showed hyperintense signals in bilateral basal ganglia, brain stem, and white matter (Figure 1). She underwent cerebrospinal fluid (CSF) analysis revealing an elevated lactate level of 3.9 mmol/L (normal: 1.1-2.3). The rest of the CSF biochemistry and cytology was within the normal range. Her blood culture was negative for any organism. She underwent detailed metabolic evaluations, including coagulation profile, serum copper, ceruloplasmin, and urinary copper, which were within normal range. Based on her clinical history and imaging and laboratory evaluations, a provisional diagnosis of adult-onset LS was made. She was managed with intravenous thiamine (300 mgm/day) with tapering dose, riboflavin, and co-enzyme-Q. Muscle biopsy from the quadricep muscle exhibited reduced cytochrome oxidase (COX) activity (Figure 2). Her muscle deoxyribonucleic acid (DNA) was extracted and analyzed for a complete mitochondrial genome, which showed no pathognomonic mutations.
Brain MRI with T2-weighted (a,b) and diffusion-weighted (c,d) images showing marked bilateral signal abnormalities in the basal ganglia, brain stem, and brain white matter.
Histochemistry from muscle fiber showing reduced cytochrome oxidase activity (a) and subsarcolemmal aggregations (b).Stains: Hematoxylin and eosin. Magnification: 160x (a), 240x (b).
She improved gradually and became conscious on day five of admission, with a gradual improvement in headache and ptosis. Her gaze improved over one and a half months, and she started ambulating over the next two months. She remained asymptomatic over subsequent follow-ups.
Discussion
LS is a progressive neurodegenerative disease of infancy and represents the most common pediatric clinical manifestation of mitochondrial disease [3]. Patients with LS typically present with episodic neurodegeneration, often leading to death at the age of three years. Although rare, adult-onset LS has also been reported, with only a few cases published. We have tabulated the reported cases of late-onset LS with clinicopathological presentation in Table 2 [4-10].
LS has remarkable clinical and genetic heterogeneity; patients with LS typically present with characteristic neuropathological features. The characteristic manifestations of LS encompass psychomotor regression or delay, limb weakness, generalized hypotonia, tremors, and lactic acidosis detected in the CSF, blood, or urine [7]. According to published reports, typical manifestations of LS may also manifest in adult-onset LS. Patients with LS manifest headaches, dementia, intellectual decline, and vertical gaze palsy based on the published data [3,6]. Hong et al. reported that patients with infantile LS manifest delayed development, motor weakness, and ataxia, and patients with late-onset LS present with vertical gaze palsy and motor weakness and ataxia. However, birth history, family history, system organ involvement, and time interval from the first clinical manifestation to LS diagnosis were not statistically significant [11]. Sakushima et al. proposed the diagnostic criteria for LS in 2011, tabulated in Table 3 [12].
Our patient presented with signs and symptoms consistent with the clinical, biochemical, and radiological criteria for LS. Muscle biopsy also reinforced the diagnosis of LS with typical imaging findings on imaging modalities. Our patient responded well to the treatment, similar to the case reported by Goldenberg and his colleagues [4].
Conclusions
Although LS typically manifests in childhood or infancy, adult-onset LS is a rare variant, with onset mainly occurring in the second or third decade of life. Diagnosis of late-onset LS exhibits significant challenges due to its diverse clinical manifestations and overlapping features with other neurological syndromes. Compared with infantile LS, late-onset LS has a favorable prognosis. Our case highlights the importance of timely recognition, genetic testing, multidisciplinary care, and individualized treatment in improving the clinical outcomes and quality of life for LS patients.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1A guide to diagnosis and treatment of Leigh syndrome J Neurol Neurosurg Psychiatry Baertling F Rodenburg RJ Schaper J 2572658520142377206010.1136/jnnp-2012-304426 · doi ↗ · pubmed ↗
- 2The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA abnormalities Ann Neurol Darin N Oldfors A Moslemi AR Holme E Tulinius M 37738349200111261513 · pubmed ↗
- 3A multicenter study on Leigh syndrome: disease course and predictors of survival Orphanet J Rare Dis Sofou K De Coo IF Isohanni P 52920142473153410.1186/1750-1172-9-52PMC 4021638 · doi ↗ · pubmed ↗
- 4Remarkable improvement in adult Leigh syndrome with partial cytochrome c oxidase deficiency Neurology Goldenberg PC Steiner RD Merkens LS 8658686020031262924910.1212/01.wnl.0000049460.72439.7f · doi ↗ · pubmed ↗
- 5Walking difficulties and brainstem dysfunction: a case report of adult onset Leigh syndrome SN Compr Clin Med Cipriano E Vecchio D Mazzini L 15452023
- 6Adult onset Leigh syndrome in the intensive care setting: a novel presentation of a C 12orf 65 related mitochondrial disease J Neuromuscul Dis Wesolowska M Gorman GS Alston CL 409419220152785875410.3233/JND-150121 PMC 5240610 · doi ↗ · pubmed ↗
- 7Adult onset Leigh syndrome Ann Indian Acad Neurol Pandit L Narayanappa G Shetty L Krishna S 55102007
- 8Late-onset Leigh syndrome without delayed development in China: a case report World J Clin Cases Liang JM Xin CJ Wang GL Wu XM 71337138920213454096910.12998/wjcc.v 9.i 24.7133 PMC 8409215 · doi ↗ · pubmed ↗