Spinal myeloid sarcoma presenting as initial symptom in acute promyelocytic leukemia with a rare cryptic PLZF::RARα fusion gene: a case report and literature review
Xuejiao Zhang, Tao Wang, Pu Chen, Yan Chen, Zhimei Wang, Tianhong Xu, Pengfei Yu, Peng Liu

TL;DR
A rare case of acute promyelocytic leukemia with a PLZF::RARα fusion gene presented as spinal myeloid sarcoma, highlighting the importance of recognizing unusual symptoms for timely diagnosis.
Contribution
Reports a rare case of APL with a cryptic PLZF::RARα fusion gene and spinal myeloid sarcoma as the initial symptom.
Findings
The patient exhibited spinal myeloid sarcoma as the first symptom of APL with a rare PLZF::RARα fusion gene.
Integrated diagnostic methods confirmed the fusion gene and APL diagnosis despite atypical presentation.
The case emphasizes the need to consider APL in patients with unexplained spinal tumors.
Abstract
Acute promyelocytic leukemia (APL) is rarely caused by the PLZF::RARα fusion gene. While APL patients with PLZF::RARα fusion commonly exhibit diverse hematologic symptoms, the presentation of myeloid sarcoma (MS) as an initial manifestation is infrequent. A 61-year-old patient was referred to our hospital with 6-month history of low back pain and difficulty walking. Before this admission, spine magnetic resonance imaging (MRI) conducted at another hospital revealed multiple abnormal signals in the left iliac bone and vertebral bodies spanning the thoracic (T11-T12), lumbar (L1-L4), and sacral (S1/S3) regions. This led to a provisional diagnosis of bone tumors with an unknown cause. On admission, complete blood count (CBC) test and peripheral blood smear revealed a slightly increased counts of monocytes. Immunohistochemical staining of both spinal and bone marrow (BM) biopsy revealed…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
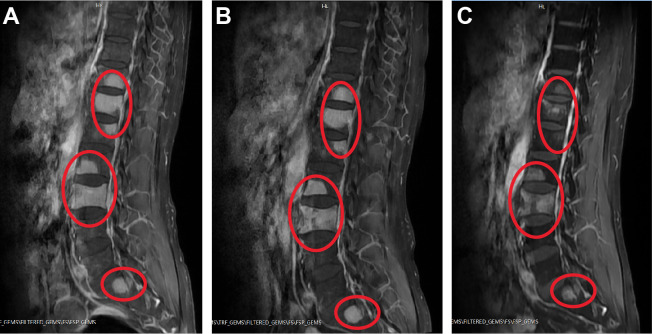 Figure 1
Figure 1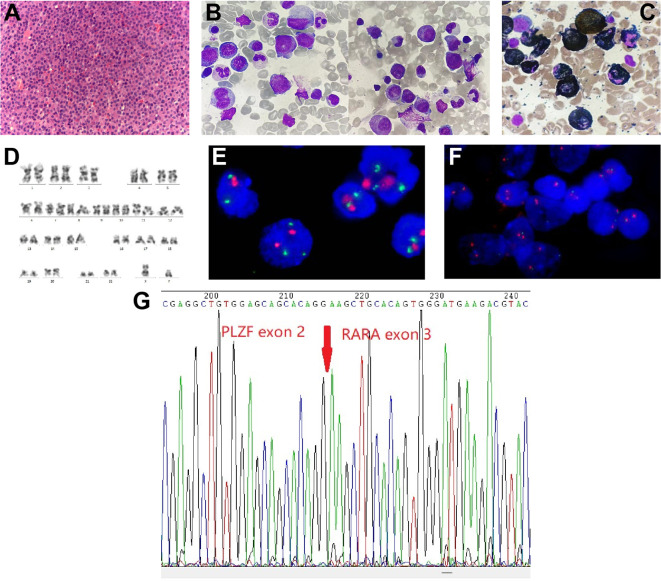 Figure 2
Figure 2| 1st cycle of therapy | 2nd cycle of therapy | normal range | |||
|---|---|---|---|---|---|
| before | after | before | after | ||
|
| 4.95 | 4.94 | 4.87 | 3.12 | 4.30-5.80 |
|
| 142 | 145 | 143 | 92 | 130-175 |
|
| 204 | 221 | 268 | 77 | 125-350 |
|
| 7.46 | 3.9 | 2.8 | 1.8 | 3.50-9.50 |
|
| 11.3 | 10.9 | 12 | 11.8 | 10.0-13.0 |
|
| 0.97 | 0.94 | 1.04 | 1.02 | 0.5-1.2 |
|
| 15.7 | 15.8 | 15 | 15.9 | 14.0-21.0 |
|
| 29.5 | 28 | 27.9 | 26.5 | 25.0-31.3 |
|
| 252 | 270 | 211 | 316 | 200-400 |
|
| 4.12 | 1.21 | 0.76 | 0.39 | 0-0.8 |
|
| 6.78 | 2.5 | 2.5 | 2.5 | <10 |
| Clinical characteristics | Treatment regimen(s) | ||||
|---|---|---|---|---|---|
| spinal MS | BM APL | other syptoms |
| ||
| 1st cycle of therapy | Yes | Yes | skin itch, rash | Yes | 1. ATRA; 2. venetoclax and azacitidine; 3. cetirizine. |
| 2nd cycle of therapy | Yes | No | pancytopenia, agranulocytosis | Yes | 1. venetoclax and idarubicin; 2. herombopag, IL-11, and blood transfusion. |
| after 2nd cycle of therapy | Yes, but in remission | No | / | No | / |
| Patient | Age/Sex | Karyotypic anomaly | Initial symptom(s) | WBC (^109/L) | Auer rods | Treatment regimen(s) | HSCT | Prognisis | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 67/male | 46,XY,t(11;17)(q23;21) | weakness, anorexia, coughing, gingival bleeding | 4.1 | No | ATRA | No | Died of pneumonia and respiratory failure (at day 20) | Chen SJ, et al. ( |
| 2 | 68/male | t(11;17)(q23.24;12.21) | NA | 10.6 | Yes | 1. Daun and Ara-C; 2. ATRA; 3. Ara-C and MTZ. | No | Died of septic shock (at month 11) | Guidez F, et al. ( |
| 3 | 81/male | 46,XY,t(11;17)(q23;21) | fatigue, dyspnea, fever, and bone pain | 7.6 | Yes | ATRA | No | Died of brain stem | Licht JD, et al. ( |
| 4 | 37/female | 46,XX,t(11;17)(q23;21) | NA | 45.2 | NA | 1. Daun and Ara-C; 2. α-interferon; 3. MTZ and ETO. | No | Died of unknown reason (at month 11) | |
| 5 | 34/male | 46,XY,t(11;17)(q23;21) | bone pain and neutropenia | 2.4 | Yes | 1. ATRA; 2. Daun and Ara-C; 3. amsacrine and Ara-C. | No | CR (4 months) | |
| 6 | 53/male | 46,XY,+(3)+(13)(q34), | spontaneous bruising | 15.3 | NA | 1. Daun and Ara-C; 2. Daun, Ara-C, and G-CSF; 3. MTZ and ETO; 4. prednisone, vincristine, 6-MP, and MTZ; 5. ATRA; 6. IDA; 7. fludarabine and Ara-C. | No | Died of congestive heart failure, DIC, and acute renal failure (at month 31) | |
| 7 | 53/male | 46,XY,t(11;17)(q23;q12-21) | fatigue, shortness of breath, spontaneous bruising | 4.5 | No | 1. ATRA, Ara-C, Daun, ETO, and G-CSF; 2. Ara-C, Daun, and ETO; 3. amsacrine, Ara-C, and ETO; 5. MTZ and Ara-C. | No | CR (10 months) | Culligan DJ, et al. ( |
| 8 | 41/male | t(11;17)(23;q21) | schizophrenia, blasts in peripheral blood | 7.8 | Yes | 1. ATRA, Ara-C, Daun, 6-MP, and PDN; 2. MTZ, Ara-C, 6-MP, and PDN. | No | Died of intracranial invasion and DIC | Hoshi S. Rinsho Ketsueki ( |
| 9 | 31/male | t(11;17)(23;q21) | hyperleukocytosis | 69 | Yes | 1. ATRA; 2.Daun and Ara-C; 2. Ara-C and amsacrine; 3. MTZ and ETO; 4. ATRA and G-CSF. | allo-HSCT | CR (51 months) | Jansen JH, et al. ( |
| 10 | 32/male | 45,X,-Y,t(11;17)(q23;q21) | NA | 11.6 | NA | 1. ATRA; 2. Daun and Ara-C. | allo-HSCT | CR (37 months) | Grimwade D, et al. ( |
| 11 | 43/male | 46,XY,i(7)(q10),t(11;17)(q23;q21) | NA | 10.4 | NA | ATRA and IDA | auto-HSCT | Died of APL replase (at month 30) | |
| 12 | 34/male | 46,XY,del(11)(q23)/45,idem,-Y/46,XY | NA | 20 | NA | 1. Daun, Ara-C, and ETO; 2. Ara-C, IDA, and ATRA. | allo-HSCT | CR (33 months) | |
| 13 | 62/male | 46,XY.ish,ins(11;17)(q23;q21,q21) | NA | 9.9 | NA | 1. Ara-C, IDA, and ETO; 2. Ara-C, ETO, gemtuzumab ozogamicin, IDA, MTZ. | No | CR (15 months) | |
| 14 | 75/male | 46,XY,t(11;17)(q23;q21)/46,idem,del(12)(p1)?/46,idem,-6,+r | NA | 2 | NA | 1. ATRA, Daun, Ara-C, and thioguanine; 2. amsacrine, Ara-C, and ETO. | No | CR (17 months) | |
| 15 | 23/male | 46,XY,t(11;17)(q23;21) | fever, bone pain (left hip and | 9.1 | NA | 1. ATO; 2. Ara-C and Daun; 3. Ara-C; 4. Ara-C and Daun. | No | CR (32 months) | George B, et al. ( |
| 16 | 83/male | 46,XY,t(11;17)(q23;21) | NA | NA | NA | 1. ATRA and Daun; 2. Daun and Ara-C; 3. ATRA, 6-MP, and MTZ. | No | CR (24 months) | Cassinat, B, et al. ( |
| 17 | 50/male | 46,XY,t(11;17)(q23;q21)/45,X,-Y,t(11;17)(q23;q21) | NA | 6.8 | NA | 1. ATRA, Ara-C, Daun, and ETO; 2. Ara-C, Daun, and ETO; 3. amsacrine, Ara-C, and ETO; 4. MTZ and Ara-C. | No | CR (73 months) | Jovanovic JV, et al. ( |
| 18 | 52/male | 46,XY,t(11;17)(q23;21) | pancytopenia | 1.62 | Yes | chemotherapy without ATRA | No | NA | Han SB, et al. ( |
| 19 | 38/female | 46,XX,t(11;17)(q23;21) | fever, dyspnea, chest pain | 23.6 | Yes | 1. ATRA, Daun, and Ara-C; 2. MTZ, ETO, and Ara-C. | No | Died of sepsis with active disease | Rohr SS, et al. ( |
| 20 | 48/male | 46,XX,t(11;17)(q23;21) | weight loss, fatigue, tonsillitis | 71.6 | Yes | 1. Daun and ATRA; 2. Daun and Ara-C; 3. Ara-C and ATO. | allo-HSCT | PR | |
| 21 | 60/female | 46,XX,der (11),der(17) | fever, dizziness, fatigue | 34 | NA | 1. ATRA and hydroxyurea; 2. ATRA, MTZ, Ara-C, and ATO; 3. ATO and chemotherapy. | No | CR (11 months) | Liu KQ, et al. ( |
| 22 | 23/male | t(11;17)(q23;q21) | fever, shortness of breath, leg swelling | NA | Yes, but few | Refuse chemotherapy | No | NA | Palta A, et al. ( |
| 23 | 49/female | 46,XX,del(5)(q13q31),t(11;17)(q23;q21) | rheumatoid arthritis, pancytopenia | 7.9 | No | 1. ATRA, IDA, Ara-C, and ETO; 2. ATRA, Ara-C, and MTZ, | allo-HSCT | CR | Piñán MA, et al. ( |
| 24 | 50/male | t(11;17)(q23;q21) | fever, knee pain | NA | No | 1. Ara-C and Daun; 2. ATRA and ATO. | No | CR | Lechevalier N, et al. ( |
| 25 | 53/male | 46,XY, t(11;17)(q23;q21) with del(5)(q22q35) | Crohn disease and macrocytic anemia | 15.4 | No | 1. ATRA; 2. Daun and Ara-C | No | NA | Dowse RT, et al. ( |
| 26 | 46/male | 46,XX,t(11;17)(q23;21) | fever, leg swelling | 35.5 | NA | 1. ATRA and ATO; 2. Ara-C and IDA; 3. MTZ, ETO, and Ara-C; 4. MTZ, Ara-C; 5. Ara-C; 6. pirarubicin and Ara-C | No | CR | Wen HX, et al. ( |
| 27 | 81/female | 46,XX,add(17)(q21) ( | back pain | NA | NA | ATRA | No | Died of pulmonary hemorrhage (at day 10) | Langabeer SE, et al. ( |
| 28 | 48/female | 46,XX,t(11;17)(q23;q21);47,idem,+22 | NA | 42.5 | NA | ATRA, hydroxycarbamide | No | Died of cerebral bleeding (at 0.3 months) | Wang XX, et al. ( |
| 29 | 44/male | 46,XY,t(11;17)(q23;21) | bone pain (lower limbs and | 52.07 | NA | ATO, Daun, and Ara-C | No | NR | |
| 30 | 52/male | 47,XY,+8/47,idem, | fever, gingival bleeding | 8.92 | NA | 1. ATRA and ATO; 2. Daun and Ara-C; 3. ATRA, Ara-C, aclarubicin, and G-CSF. | No | CR (7 months) | |
| 31 | 62/male | 46,XY,t(11;17)(q23;21) | gout, pancytopenia | 2.99 | Yes, but few | 1. Ara-C and IDA; 2. Ara-C, IDA, and ATRA; 3. Ara-C and ATRA | No | CR | Pardo Gambarte L, et al. ( |
| 32 | 51/male | 46,XY,t(11;17)(q23;q21) | fatigue, easy bruising | NA | NA | NA | No | NA | Liu G, et al. ( |
| 33 | 56/male | t(11;17)(q23;q21) | apnoea, night sweats | 25.47 | No | IDA and Ara-C | allo-HSCT | CR (2 years) | Canali A and Rieu JB ( |
| 34 | 44/male | 45,X,-Y,t(11;17)(q23;q21) | flu-like illness | NA | No | fludarabine, Ara-C, G-CSF, IDA, and venetoclax. | No | CR | Courville EL, et al. ( |
| 35 | 56/male | 46,XY,add (9)(q11) | lower extremity paralysis | 7.1 | No | NA | NA | NA | Cho EJ, et al. ( |
| 36 | 66/male | t(11;17)(q23;q21) | fever, weight loss, arthralgia | 11.1 | No | 1. steroids; 2. Ara-C and Daun | allo-HSCT | CR (1.5 years) | Castelijn DAR, et al. ( |
| 37 | 15/male | NA | abdominal pain, weakness, fever | 64.94 | No | 1. IDA and ATO; 2. Ara-C; 3. ATRA | No | CR | Rabade N, et al. ( |
| 38 | 38/male | NA | easy fatigability, dyspnea, and fever | NA | Yes | ATO | No | Died of unknown reseaon (at 2 months) | |
| 39 | 45/male | NA | easy fatigability and fever | NA | No | ATO | No | NA | |
| 40 | 36/male | NA | fever and rash | 4.86 | No | 1. decitabine and ATO; 2. Daun and Ara-C; 3. Ara-C | No | Death in relapse (at 11 months) | |
| 41 | 22/male | NA | fever and body ache | 76.99 | No | 1. ATO, Daun, and Ara-C; 2. Ara-C | No | NA |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRetinoids in leukemia and cellular processes · Acute Myeloid Leukemia Research · Histone Deacetylase Inhibitors Research
Introduction
Acute promyelocytic leukemia (APL), also known as acute myeloid leukemia (AML) subtype M3 according to the French-American-British (FAB) classification, is primarily characterized by an accumulation of immature promyelocytes in bone marrow (BM) (1). APL patients typically appear as one or more of hematologic symptoms, including fever, bleeding, fatigue, infections, bone pain, and others (1). Besides, in some cases, APL may present with extramedullary involvement that causes myeloid sarcoma (MS) (2–6). Although rare in clinical practice, MS is more commonly associated with relapsed or refractory APL cases, with an estimated incidence of 3%–5% (2). However, in newly diagnosed APL, MS occurs even more rarely, potentially contributing to delays in APL diagnosis (3–6). In addition, MS can occur simultaneously in various extramedullary locations, such as skin, soft tissues, bones, lymph nodes, and other organs (3–6). Therefore, MS may produce miscellaneous non-hematologic symptoms that mimic those of other diseases, making it more challenging to timely distinguish MS. Moreover, the atypical morphological characteristics exhibited by leukemia cells at onset of this disease adds complexity to the diagnostic procedure in cases of APL with MS (4–6). Given the substantial risk of disseminated intravascular coagulation (DIC) in association with APL, a condition that can be severe and life-threatening, it is imperative to prioritize early APL diagnosis and the immediate commencement of APL-specific treatments like all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) (1, 7, 8).
One of the key diagnostic features of APL is chromosomal translocation involving the gene that encodes retinoic acid receptor alpha (RARα) on chromosome 17 (1, 9–11). In particular, an overwhelming majority of APL cases exhibit the typical t(15;17)(q22;21) translocation, which results in the fusion of the promyelocytic leukemia (PML) gene and RARα gene, namely PML::RARα fusion gene (9). In exceptionally infrequent cases (1~2%), APL has been observed with rare variant translocations, including t(11;17)(q23;q21), t(11;17)(q13;q21), t(5;17)(q32;q21), and t(17;17)(q11;q21) (10, 11). These variant translocations involve other partner genes and may impact on the clinicopathologic features of APL. For example, APL patients with the classic PML::RARα fusion gene are highly responsive to ATRA and ATO (1, 9). However, patients with the t(11;17)(q23;q21) translocation, resulting in the fusion of the promyelocytic leukemia zinc finger (PLZF)-encoding gene and RARα gene—a fusion less prevalent than PML::RARα—may display a comparatively less robust response to the same treatments (10–12). It is important to note that some thirty APL cases with MS as initial presentation has been documented in literatures so far, and almost all carried the classic t(15;17)(q22;21) translocation (3–6). The development of MS is still scarcely reported in APL with other rare variant translocations. Here, we report a newly diagnosed APL patient (61-year-old male) with spinal MS as the first presentation. Using integrated genetic testing, we identified a normal karyotype, and notably, a rare cryptic PLZF::RARα fusion gene.
Case presentation
A 61-year-old Chinese man was referred to our hospital with 6-month history of low back pain and difficulty walking, which were particularly severe after physical exertion. The patient reported occasional temporary relief of these symptoms through Chinese medical massage treatments. One month before being admitted, his symptoms had worsened progressively without a clear precipitating factor, and he experienced pain that extended to his left thigh accompanied by a sensation of numbness. Subsequently, detailed bone examinations were performed at local hospital. At that time, spine magnetic resonance imaging (MRI) suggested pathologic fracture of lumbar (L3) spine and showed multiple abnormal signals in left iliac bone and in vertebral bodies of the thoracic (T11-T12), lumbar (L1-L4), and sacral (S1/S3) spine. Meanwhile, fluorine-18-fluorodeoxyglucose (FDG) positron emission tomography/computed tomography (PET/CT) imaging reported that FDG uptake was slightly increased in those bone lesions, but not in other areas of the body. The patient was tentatively diagnosed with bone tumors of unknown cause and was transferred to our hospital for further diagnosis and treatment.
On admission, our spine MRI confirmed the previous results (Figure 1A). A complete blood count (CBC) test showed a slightly increased counts of monocytes (0.73×10^9^/L; normal range: 0.1-0.6×10^9^/L), and normal results of red blood cells (4.95×10^12^/L), hemoglobin (142 g/L), platelets (204×10^9^/L), and white blood cells (7.46×10^9^/L) (Table 1). Examination of the peripheral blood smear consistently suggested an increased proportion of monocytes. Moreover, antibody serology tests revealed positive results for antinuclear (ANA) antibody and anti-beta-2 glycoprotein 1 (B2GP1) antibody. With flow cytometry (Becton Dickinson FastImmune™ Cytokine System), we detected elevated levels of interferon gamma (IFN-γ; 13.03 pg/mL; normal range:≤4.43 pg/mL) and interleukin-17A (5.60 pg/mL; normal range:≤4.74 pg/mL) in whole blood. Notably, coagulation tests revealed a significant increase in D-dimers level (4.12 mg/L; normal range: 0-0.8 mg/L), while other coagulation indexes were within normal range (Table 1). Heart, renal, liver, and nervous system functions were normal.
Examples of spinal sagittal T1-weighted MRI of the patient. (A) On admission. (B) After first cycle of therapy. (C) After second cycle of therapy. Abnormal high signals on T1 images are shown circled in red.
In order to assess the histopathological basis of bone lesions, we further performed CT-guided percutaneous needle biopsy of lumbar L3. The spinal biopsy results indicated the presence of immature/blast-like cells with eccentric nuclei within the spaces of the bone trabeculae (Figure 2A). Immunohistochemical staining of the spinal biopsy revealed positive expression for CD117, CD43, myeloperoxidase (MPO), lysozyme, and Ki67 (labelling index about 40%), while it tested negative for CD20, CD34, CD56, CD61, CD79a, CD138, and IgG/M, κ, λ expression. These findings suggested the presence of myeloid neoplasms. Meanwhile, BM biopsy revealed 95% of blast cells and a staining profile characterized by CD117 (+), MPO (+) and lysozyme (part+), which was similar to the results of spinal biopsy. In addition, BM aspirate showed hypercellularity with an elevated myeloid/erythroid (M/E) ratio of 7.52:1. Specifically, there was a significant elevation in the percentage of promyelocytes (21%; normal range: 0.4-3.9%), strongly indicating the likelihood of APL. Erythropoiesis was insufficient, while megakaryopoiesis was normal. Giemsa-stained promyelocytes displayed round nuclei and hypergranular cytoplasm (Figure 2B). However, Auer rods were notably absent. The majority of promyelocytes had positive staining for MPO (Figure 2C). Multiparameter flow cytometry of BM aspirate detected 78% blasts and suggested an immunophenotype that was positive for CD13, CD33, CD117, and MPO, and negative for CD3, CD10, CD11b, CD14, CD15, CD19, CD34, CD71, CD79a, and HLA-DR, corresponding to APL features.
Spine tissue and bone marrow (BM) aspirate examinations and results of karyotype and fluorescence in situ hybridization (FISH) analysis. (A) Histopathology of spinal cord (hematoxylin and eosin staining; magnification, ×400). (B) BM aspirate shows atypical acute promyelocytic leukemia morphology (Wright-Giemsa staining; magnification, ×1,000). (C) BM aspirate showed that promyelocytes were myeloperoxidase (MPO) positivity (MPO staining; magnification, ×1,000). (D) Chromosome analysis of the BM reveals a normal male karyotype. (E, F) Metaphase FISH analysis of the patient. (E) The standard dual-color, dual-fusion probe set probe set for t(15;17) shows the presence of two red signals (PML) and three green signals (RARα), suggesting RARα rearrangement; (F) The RARα break-apart probe detects separated red signals (RARα). (G) Sanger sequencing analysis demonstrates PLZF::RARα fusion transcript.
Notably, cytogenetics G-band analysis of BM cells revealed a normal male karyotype (46, XY) (Figure 2D). Metaphase fluorescence in situ hybridization (FISH) analysis with the PML::RARα dual-color dual-fusion probe kit (FP-005, Wuhan HealthCare Biotechnology Co., Ltd.) on BM aspirate suggested the absence of PML-RARα dual-fusion translocation (Figure 2E). However, three green FISH signals suggested the presence of RARα translocation (Figure 2E). This finding was subsequently validated using the RARα break-apart probe detection kit (FP-043, Wuhan HealthCare Biotechnology Co., Ltd.) (Figure 2F). To further explore the etiology, we performed reverse transcriptase polymerase chain reaction (RT-PCR) (Dian Diagnostics Group Co. Ltd., Hangzhou, China) on BM. It revealed PLZF::RARα fusion by using the reverse primers (NM_000964; RARα 1-R, 5’-AAGCCCTTGCAGCCCTCAC-3’ [external]; RARα 2-R, 5’-CCCATAGTGGTAGCCTGAGGAC-3’ [internal]) located within exon 2 of RARα gene in conjunction with the forward primers (NM_001018011; PLZF 1-F, 5’-CCACAAGGCTGACGCTGTATT-3’ [external]; PLZF 2-F, 5’-GTGGGCATGAAGTCAGAGAGC-3’ [internal]) located within exon 3 of PLZF gene. Sanger sequencing further confirmed the presence of PLZF::RARα exon 3–exon 2 fusion transcript (Figure 2G). Next-generation sequencing (NGS) analysis with the Myeloid Tumor Assay that was consisted of 128 genes panel (Dian Diagnostics Group Co., Hangzhou, China) detected no additional mutations. Taken together, according to FAB classification, a definitive diagnosis of APL was ultimately established.
In the initial induction therapy, the patient was treated with 20 mg/day ATRA (BID) for one week. This was followed by a regimen incorporating subcutaneous azacitidine (120 mg/day, Day 1 to 7) and oral administration of venetoclax with a progressive dose escalation: 100 mg/day (Day 1), 200 mg/day (Day 2), and 400 mg/day (Day 3 to 24) (Table 2). During this period, the patient was also treated with cetirizine for skin itch and rash. Subsequent CBC revealed that his WBC counts reduced to 3.9×10^9^/L, which was still within normal range (Table 1). BM aspirate showed hypercellularity and a decreased M/E ratio of 0.2:1, which was characterized by granulocytic hypoplasia and erythrocytic/megakaryocytic hyperplasia. Importantly, BM aspirate indicated that the percentage of promyelocytes reduced to 0.5%. On BM biopsy, residual leukemia cells were negligible. However, spine MRI showed no significant improvement in MS lesions (Figure 1B). RT-PCR from BM showed the persistence of PLZF::RARα fusion.
As a result, we maintained the patient on oral venetoclax administration at 400 mg/day (Day 1 to 12) and further administered idarubicin intravenously (10 mg/day IV bolus, Day 1 and 2; 20 mg/day IV bolus, Day 3) (Table 2). Meanwhile, the patient developed pancytopenia, and had sustained agranulocytosis for two weeks. To address this, herombopag, recombinant human interleukin-11 (IL-11), and blood transfusion were given (Table 2). Repeated BM aspirate showed reduced cellularity and a decrease in all three blood cell lineages. Notably, the percentage of promyelocytes increased again to 12%, but subsequent flow cytometry immunophenotyping confirmed a normal phenotype of immature granulocytes, which was hypothesized to be a possible manifestation of myeloid hematopoietic recovery. Fortunately, MRI showed that spinal MS lesions were obviously shrunken (Figure 1C). The patient also obtained symptomatic relief of low back pain and difficulty walking. What’s more important, PLZF::RARα fusion transcript became undetectable, indicating the achievement of complete molecular remission (MR). The decision to initiate additional treatment was contingent upon the successful recovery of the patient’s hematopoietic functions.
Discussion
According to the new International Consensus Classification (ICC) of myeloid neoplasms and acute leukemias, APL with t(11;17)(q23;q21) translocation is now redefined as APL with other RARα rearrangements (13). Since the first report in 1993, only about forty newly diagnosed APL patients with t(11;17)(q23;q21) have been documented in literatures (Table 3). This rare APL impacts individuals across a broad spectrum of ages, ranging from 15 to 81 years old, with an average age of 48.8 years (Table 3). Interestingly, the prevalence of APL with t(11;17)(q23;q21) appears to be higher in males (35/41; 85.4%) compared to females (6/41; 14.6%) (Table 3). The t(11;17)(q23;q21) translocation gives rise to PLZF::RARα fusion gene, also referred to as ZBTB16::RARα. PLZF exhibits the ability to bind to DNA, thereby governing the transcriptional activity of genes pivotal to diverse cellular functions, particularly those involved in the differentiation and maturation of promyelocytic cells (40). However, it’s important to highlight that, in very rare APL cases, the karyotype may appear normal, and the fusion gene may be formed through cryptic or subtle rearrangements that are not readily detected by standard cytogenetic analysis (19, 41). Similar to our patient, Grimwade D et al. previously reported an APL case with a normal karyotype and cryptic formation of the PLZF::RARα fusion gene (19). Meanwhile, studies suggested that cryptic formation was not only limited to PLZF::RARα, but also identified in APL with PML::RARα (41, 42), IRF2BP2::RARα (43), TBL1XR1::RARα (44), and FIP1L1::RARα (45). Such exceptional APL cases underscore the critical importance of employing molecular techniques, such as FISH or RT-PCR, to pinpoint the precise genetic abnormality and confirm the final diagnosis of APL.
Moreover, it’s noteworthy that a majority of APL patients harboring the PLZF::RARα fusion initially manifest with non-specific symptoms that were identical to classical APL, including fever, pancytopenia, fatigue, bone pain, and so forth (Table 3). MS is generally considered a rare extramedullary manifestation of untreated APL, but after induction therapy MS becomes more common (2, 3). As of our current information, our patient was actually the second report of APL with PLZF::RARα fusion and MS as the initial symptom. The previous case was a 56-year-old Korean man characterized by APL and spinal MS (37). Even in classical APL, only around thirty cases with MS have been reported thus far (3–6). In addition, a recent report by Wang, Y., et al. highlighted the presence of skull MS in a 28-month-old girl with APL caused by FIP1L1::RARα fusion (45). The fact that MS has been identified in APL with different variants suggests that MS may not be exclusive to a particular genetic fusion. However, the exact mechanism underlying the development of MS in APL is not fully understood, and it may involve various processes related to the behavior of leukemia cells. In patients with AML or APL, MS can manifest in various sites throughout the body. Bone represents a frequent site of involvement, with MS lesions often observed in spine, skull and long bones (4, 6, 45–47). Additionally, soft tissues including skin, subcutaneous tissue, and lymph nodes are susceptible to MS infiltration (2, 6, 48). In more severe cases, MS can affect visceral organs such as liver, colon, and central nervous system (CNS) (5, 49, 50). The presentation of MS varies widely based on the affected site(s), necessitating a comprehensive diagnostic approach and tailored treatment strategies.
Morphological characteristics of abnormal promyelocytes exhibit variability among APL patients with the PLZF::RARα fusion, occasionally differing significantly from those seen in classic APL (11, 29, 33, 51). In classic APL, distinguishing features of promyelocytes encompass lobulated nuclei, hypergranular cytoplasm, and Auer rods (1, 51). However, a subgroup of APL patients with the PLZF::RARα fusion, similar to our patient, may present with atypical traits, such as round/non-lobulated nuclei, hypogranular or entirely agranular cytoplasm, along with the absence of Auer rods (Table 3). Notably, studies have found that APL cases with the PLZF::RARα fusion may exhibit vacuoles or square crystalline structures within the cytoplasm of promyelocytes (29, 33). Interestingly, we also observed small vacuoles in few abnormal promyelocytes from our patient. Further research is needed to better understand the underlying mechanisms leading to the formation of these atypical intracytoplasmic inclusions and their clinical significance. Hence, in instances with atypical presentations, the use of stains like MPO, Sudan Black B, and immunohistochemical markers such as CD13, CD33, and CD117 can be valuable in reinforcing the diagnosis of APL (1, 13). Nevertheless, it should be noted that APL patients may infrequently show negativity for both MPO and Sudan Black B staining (52, 53), and the immunophenotype may also undergo changes after induction therapy (54).
The immediate initiation of ATRA is now a crucial element in the induction therapy for classic APL, resulting in a notable rise in complete remission (CR) rates and enhanced overall outcomes (8, 9). Currently, there is no established consensus guideline regarding the utilization of ATRA in the treatment of APL with rare variants and MS. Despite demonstrating the ability of leukemia cells carrying the PLZF::RARα fusion to fully differentiate with both ex vivo and in vivo ATRA treatment, the clinical reality is that APL with this rare fusion is commonly considered ATRA-insensitive and is linked to an unfavorable prognosis (10–12, 55). Significantly, it’s also been reported that a small number of APL patients with PLZF::RARα fusion who underwent a combination of ATRA and intensive chemotherapy achieved CR (11, 33). In recent years, the BCL-2 inhibitor venetoclax has exhibited encouraging therapeutic outcomes in AML as well as other hematological malignancies (56). Interestingly, exploratory studies suggested that APL patients who are resistant to conventional chemotherapies may derive benefit from regimens incorporating venetoclax (57). In particular, APL patients harboring exceedingly uncommon RARα::HNRNPC and RARα::THRAP3 fusions have been documented to achieve CR through the administration of venetoclax and hypomethylating agents such as azacytidine or decitabine (58, 59). These findings prompted us to initiate treatment with ATRA, followed by a combination of venetoclax and azacytidine in our patient. The treatment demonstrated a significant efficacy in eradicating leukemic cells from BM aspirate; however, its impact on alleviating his MS and achieving MR was negligible. Fortunately, the substitution of azacitidine with the anthracycline antineoplastic agent idarubicin has ultimately led to the achievement of MR, albeit the occurrence of significant hematological toxicity.
Conclusions
To summarize, we report the clinical features and outcome of a rare APL patient characterized by a cryptic PLZF::RARα fusion and MS as the initial presenting symptom. Our study not only offers valuable insights into the heterogeneity of APL clinical manifestations but also emphasizes the crucial need to promptly consider the potential link between APL and MS for ensuring a timely diagnosis and personalized treatments.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by the ethics committee of Zhongshan Hospital of Fudan University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
XZ: Data curation, Investigation, Writing – original draft. TW: Formal analysis, Investigation, Writing – review & editing. PC: Formal analysis, Writing – review & editing. YC: Formal analysis, Writing – review & editing. ZW: Project administration, Writing – review & editing. TX: Investigation, Writing – review & editing. PY: Investigation, Writing – review & editing. PL: Funding acquisition, Supervision, Writing – review & editing.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Cingam SR Koshy NV. Acute Promyelocytic Leukemia. In: Stat Pearls. Stat Pearls Publishing, Treasure Island (FL (2023).29083825 · pubmed ↗
- 2Vega-Ruiz A Faderl S Estrov Z Pierce S Cortes J Kantarjian H. Incidence of extramedullary disease in patients with acute promyelocytic leukemia: a single-institution experience. Int J Hematol. (2009) 89:489–96. doi: 10.1007/s 12185-009-0291-8 PMC 419930219340529 · doi ↗ · pubmed ↗
- 3Kasinathan G Sathar J. Extramedullary disease in acute promyelocytic leukaemia: A rare presentation. SAGE Open Med Case Rep. (2020) 8:2050313 X 20926076. doi: 10.1177/2050313 X 20926076 PMC 726815632537161 · doi ↗ · pubmed ↗
- 4Shu X Wu Q Guo T Yin H Liu J. Acute promyelocytic leukemia presenting with a myeloid sarcoma of the spine: A case report and literature review. Front Oncol. (2022) 12:851406. doi: 10.3389/fonc.2022.851406 35311073 PMC 8931201 · doi ↗ · pubmed ↗
- 5Wang L Cai DL Lin N. Myeloid sarcoma of the colon as initial presentation in acute promyelocytic leukemia: A case report and review of the literature. World J Clin Cases. (2021) 9:6017–25. doi: 10.12998/wjcc.v 9.i 21.6017 PMC 831696334368322 · doi ↗ · pubmed ↗
- 6Harrer DCLüke F Einspieler I Menhart K Hellwig D Utpatel K. Case report: extramedullary acute promyelocytic leukemia: an unusual case and mini-review of the literature. Front Oncol. (2022) 12:886436. doi: 10.3389/fonc.2022.886436 35692786 PMC 9174987 · doi ↗ · pubmed ↗
- 7Dombret H Scrobohaci ML Ghorra P Zini JM Daniel MT Castaigne S. Coagulation disorders associated with acute promyelocytic leukemia: corrective effect of all-trans retinoic acid treatment. Leukemia. (1993) 7:2–9.8418375 · pubmed ↗
- 8Osman AEG Anderson J Churpek JE Christ TN Curran E Godley LA. Treatment of acute promyelocytic leukemia in adults. J Oncol Pract. (2018) 14:649–57. doi: 10.1200/JOP.18.00328 30423270 · doi ↗ · pubmed ↗