Interaction between miR-142-3p and BDNF Val/Met Polymorphism Regulates Multiple Sclerosis Severity
Ettore Dolcetti, Alessandra Musella, Sara Balletta, Luana Gilio, Antonio Bruno, Mario Stampanoni Bassi, Gianluca Lauritano, Fabio Buttari, Diego Fresegna, Alice Tartacca, Fabrizio Mariani, Federica Palmerio, Valentina Rovella, Rosangela Ferese, Stefano Gambardella

TL;DR
This study shows how a genetic variation in the BDNF gene interacts with miR-142-3p to influence the severity of multiple sclerosis.
Contribution
First investigation of how BDNF Val66Met polymorphism affects miR-142-3p's role in MS severity.
Findings
Met-carrier patients showed a decoupling of miR-142-3p levels from IL1β levels in cerebrospinal fluid.
BDNF Val66Met polymorphism interferes with the IL1β-miR-142-3p axis, affecting MS progression and severity.
Findings suggest personalized medicine approaches could be improved by considering this genetic interaction.
Abstract
MiR-142-3p has recently emerged as key factor in tailoring personalized treatments for multiple sclerosis (MS), a chronic autoimmune demyelinating disease of the central nervous system (CNS) with heterogeneous pathophysiology and an unpredictable course. With its involvement in a detrimental regulatory axis with interleukin-1beta (IL1β), miR-142-3p orchestrates excitotoxic synaptic alterations that significantly impact both MS progression and therapeutic outcomes. In this study, we investigated for the first time the influence of individual genetic variability on the miR-142-3p excitotoxic effect in MS. We specifically focused on the single-nucleotide polymorphism Val66Met (rs6265) of the brain-derived neurotrophic factor (BDNF) gene, known for its crucial role in CNS functioning. We assessed the levels of miR-142-3p and IL1β in cerebrospinal fluid (CSF) obtained from a cohort of 114…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
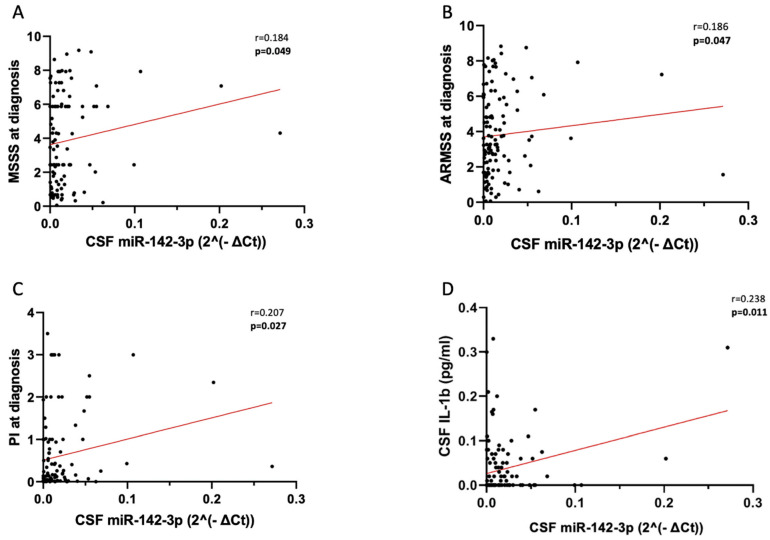 Figure 1
Figure 1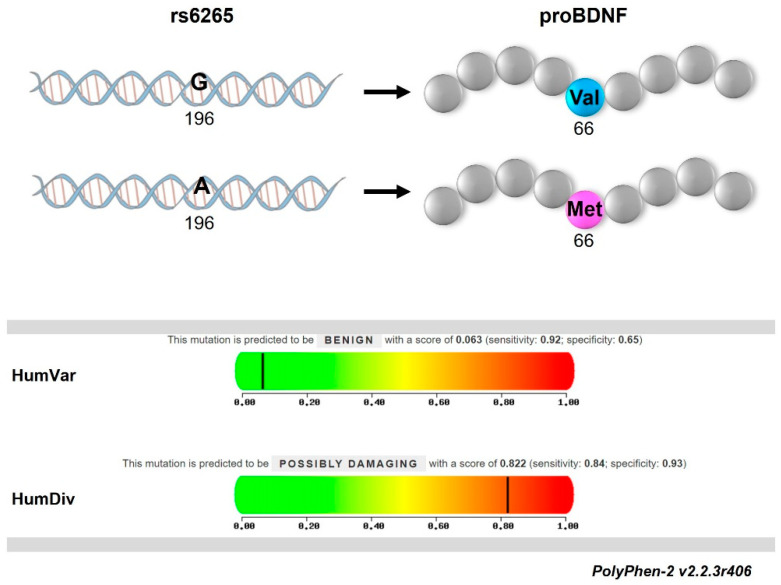 Figure 2
Figure 2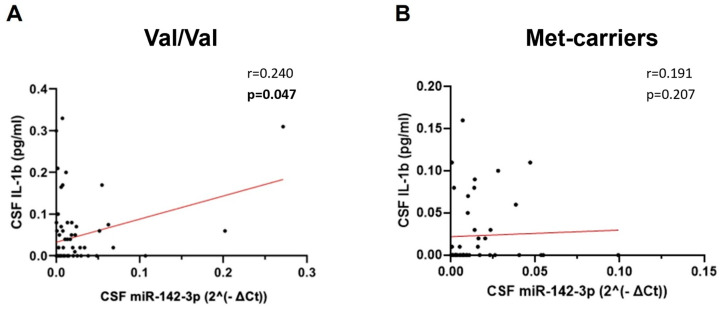 Figure 3
Figure 3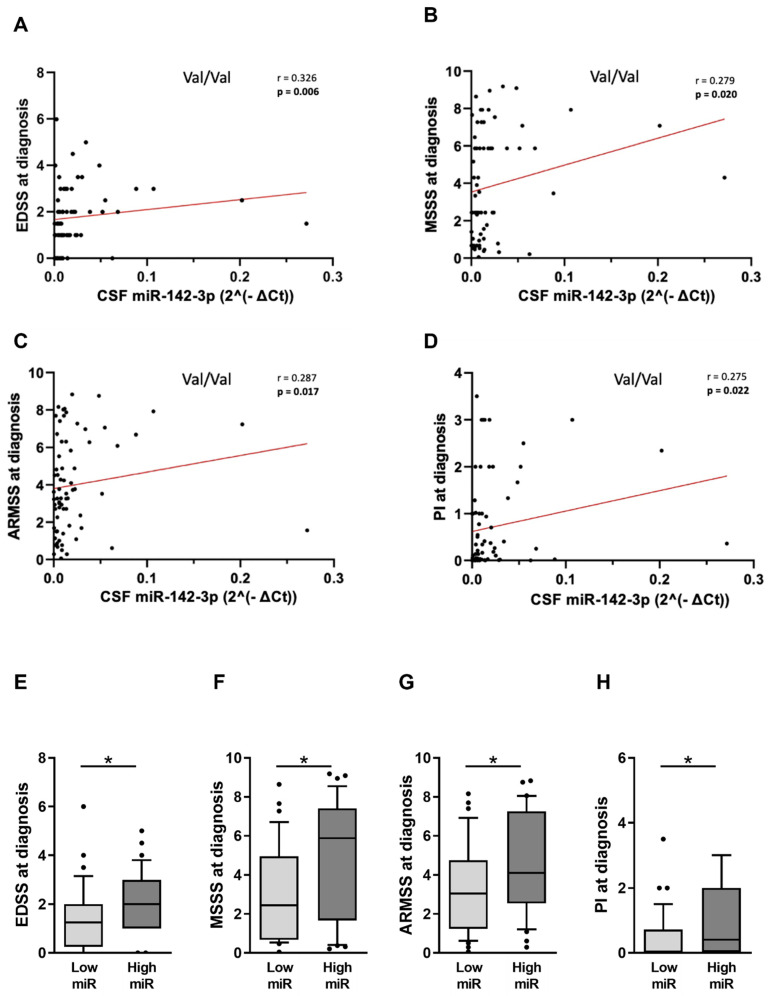 Figure 4
Figure 4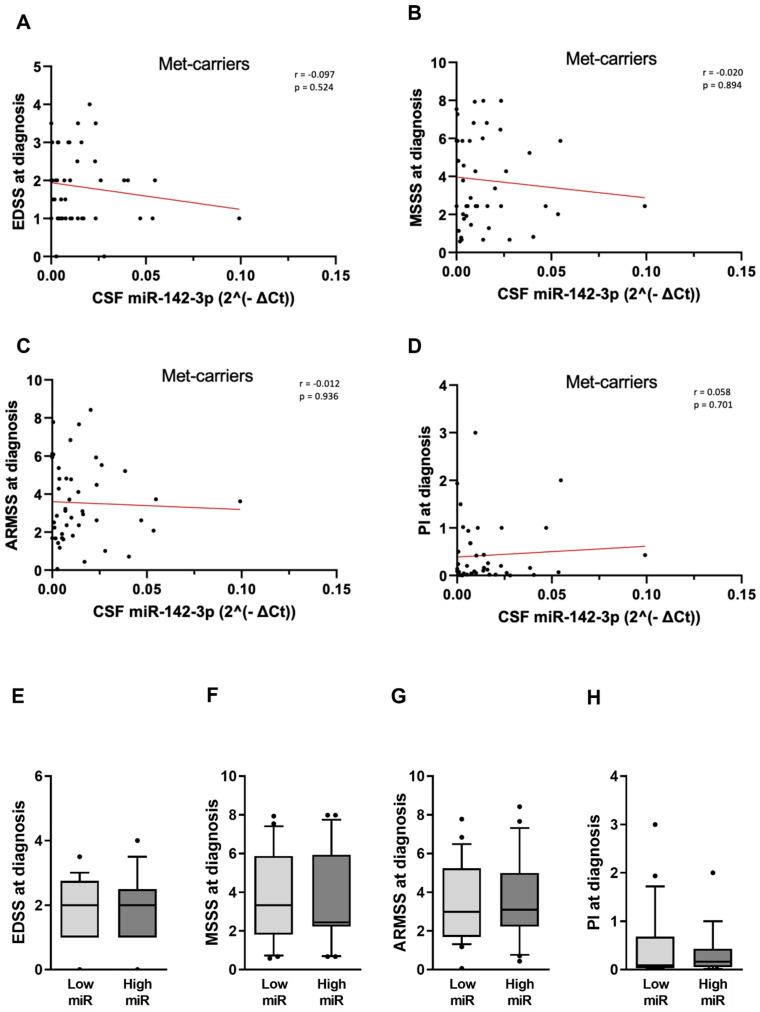 Figure 5
Figure 5- —Italian Ministry of Health
- —Ministry of University and Research (MUR), National Recovery and Resilience Plan (NRRP)
- —Fondazione Italiana Sclerosi Multipla (FISM) and financed or co-financed with the ‘5 per mille’ public funding
- —Private donations
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsOrganizational Management and Innovation · Business, Education, Mathematics Research · Diverse Applied Research Studies
1. Introduction
MS represents one of the most common autoimmune disabling diseases of the CNS, characterized by synaptic damage, demyelination, and early neuronal death mediated by neuroinflammation [1,2]. MS manifests in various clinical forms, with the relapsing-remitting MS (RRMS) being the most common [1]. The pathophysiology of RRMS involves varying degrees of acute and chronic inflammation and neurodegeneration, and the intricate interplay of genetic and epigenetic factors contributing to the heterogeneity of the disease [3]. Patients with RRMS (pwRRMS) experience alternating periods of clinical manifestations and remission, eventually transitioning into the progressive form marked by irreversible and advancing disability [1]. Many investigations have indicated that excitotoxic synaptic damage can contribute to the silent MS progression [2,4].
The molecular triggers of MS synaptic alterations include not only proinflammatory cytokines but also microRNAs (miRNA). Among these post-transcriptional regulators of gene expression, miR-142-3p stands up for importance in MS excitotoxicity [5,6,7]. The dual nature of miR-142-3p has clearly emerged due to its action on both immune and synaptic compartments. Its role in modulating immune responses and neuroinflammation has been well established for many years [8,9]. More recently, MS preclinical and clinical studies have revealed that miR-142-3p is part of a detrimental regulatory axis with the pro-inflammatory cytokine IL-1β, which contributes to excitotoxic synaptic overstimulation with serious consequences for disease prognosis and the efficacy of disease-modifying treatments [5,6]. Compelling evidence from a chimeric ex vivo MS model showed that high levels of miR-142-3p in the human CSF can impair synaptic transmission in healthy murine brain slices, mimicking synaptic damage typically observed in experimental MS [5].
However, the complete understanding of the molecular interactors of miR-142-3p in the MS excitotoxic pathway and the influence of individual genetic variability on miR-142-3p action remains elusive. Notably, miR-142-3p has been recently demonstrated to indirectly affect the transcription of Bdnf by targeting the calmodulin-kinase 2 (Camk2a) mRNA in in vitro activated microglia [10].
BDNF is a pivotal neurotrophin that has garnered substantial attention in MS also due to its potential implications in inflammatory synaptic dysfunctions [11].
The BDNF gene, located on chromosome 11 in humans [12], produces many transcripts by combining nine exons, each with its own promoter triggered by different stimuli [13]. Each mRNA isoform consists of two exons and encodes for the same BDNF protein, but their abundance varies across different brain regions. Additionally, the primary BDNF transcripts can be processed at two alternative polyadenylation sites, resulting in mature mRNAs with short or long 3′UTRs of different lengths [14,15]. Neuronal activity heavily influences most of the regulatory mechanisms controlling BDNF expression [16]. Furthermore, BDNF is initially synthesized as the proBDNF precursor protein, which is cleaved by intracellular or extracellular peptidase enzymes into mature BDNF and propeptide [17].
BDNF secretion at the synapsis is altered in neuroinflammation [18] and its action is dependent from single-nucleotide polymorphisms (SNPs), such as BDNF Val66Met (rs6265). This SNP is in the last exon, present in all BDNF transcripts, and represents the most extensively studied SNP in the BDNF gene. The substitution of a valine (Val) to methionine (Met) at position 66 of the proBDNF protein has been associated with numerous impaired functions of the CNS, moving from neuronal survival and synaptic plasticity to the modulation of immunophenotypes in resident and non-resident cells [19,20,21,22,23].
Our study aims to evaluate the clinical significance of the interaction between CSF miR-142-3p levels and the Val66Met polymorphism in pwRRMS at diagnosis, considering its potential prognostic significance.
2. Results
2.1. CSF miR-142-3p Associates with Adverse Clinical Signs of RRMS
In order to assess the clinical relevance of miR-142-3p in the early phases of MS, we evaluated its CSF levels at diagnosis in a cohort of 114 pwRRMS (Table 1) and explored its associations with clinical and demographical characteristics.
We observed a significant positive correlation between CSF miR-142-3p levels, and key indicators of disease severity, like MSSS (Figure 1A: Spearman’s Rho = 0.184, p = 0.049) and ARMSS (Figure 1B: Spearman’s Rho = 0.186, p = 0.047). The levels of miR-142-3p in the CSF were also correlated with disease progression, as estimated by PI (Figure 1C: Spearman’s Rho 0.207, p = 0.027). No significant associations with other clinical and demographical parameters were found (age at diagnosis: p = 0.335, sex p = 0.063, disease duration: p = 0.441, EDSS p = 0.061, radiological activity p = 0.371, presence of OCB p = 0.807).
Moreover, we confirm the presence of the detrimental regulatory axis IL-1β-miR-142-3p in the CSF of pwRRMS, as evidenced by a direct correlation between the levels of these two excitotoxic molecules in the biofluid (Figure 1D: Spearman’s Rho = 0.238, p = 0.011), consistent with previous findings established in a more heterogenous cohort of patients [6].
2.2. Met66-Allele Is Not Associated with the Risk of RRMS Onset but Can Contribute to Disease Course Heterogeneity
The genetic screening of pwRRMS participating in our study for the SNP BDNF Val66Met revealed an aligned distribution of Val-allele (frequency of G = 78.51%) and Met-allele (frequency of A = 21.49%) with general Caucasian population (chi-square n.s. p = 0.499). Genotype frequencies were in Hardy–Weinberg equilibrium (chi-square n.s. p = 0.695) with 60.52% Val66 homozygotes (GG, n = 69), 35.96% heterozygotes (AG, n = 41) and 3.50% Met66 homozygotes (AA, n = 4). Coherently, the pathogenicity prediction of Val66Met substitution by PolyPhen-2 algorithm (Figure 2) indicated no drastic effects of the SNP on the risk of diseases from all the remaining genetic variations (HumVar score = 0.06; sensitivity = 0.92; specificity = 0.65) but suggested its involvement in the endophenotypes of multifactorial diseases such as MS (HumDiv score = 0.82; sensitivity = 0.84; specificity = 0.93).
2.3. The IL1β-miR-142-3p Axis Is Disrupted in pwRRMS Carrying Met-Allele
The well-established synaptic effect of Val66Met substitution in proBDNF [19,20] and the recently discovered link between BDNF and miR-142-3p in neuroinflammation [10] prompted us to investigate the SNP influence on the excitotoxic IL1β-miR-142-3p axis in the CSF of pwRRMS. To this aim, we examined separately the Val66 homozygotes (Val/Val; Table 2) and the Met-carriers (Val/Met and Met/Met; Table 3) present in our cohort. These two groups of patients were equivalent across all demographic and clinical characteristics assessed at diagnosis, with exception of age (Val/Val, median [IQR] = 31.3 [25.1–41.7], Met-allele carriers, median [IQR]: = 39.8 [31.5–47.2], Mann–Whitney test p = 0.010). CSF values of miR-142-3p and IL-1β were unchanged in the two conditions (CSF miR-142-3p: Val/Val median [IQR] = 0.009 [0.004–0.020], Met-allele median [IQR] = 0.009 [0.003–0.020], Mann–Whitney test p = 0.683; CSF IL-1β: Val/Val median [IQR] = 0.0002 [0.000–0.060], Met-allele median [IQR] = 0.0001 [0.000–0.030], Mann–Whitney test p = 0.209).
In Val/Val patients (Table 2), we observed a significant direct correlation between the CSF levels of miR-142-3p and IL-1β (Figure 3A; Spearman’s Rho = 0.240, p = 0.047, n = 69), as there was in the whole cohort of patients (Figure 1D). Conversely, the association between these molecules was lost in the Met-carrier group (Table 3; Figure 3B; Spearman’s Rho = 0.191, p = 0.207, n = 69 45), indicating the disruption of the regulatory axis IL1β-miR-142-3p.
2.4. The Detrimental Effect of miR-142-3p on Clinical Parameters Is Impaired in pwRRMS Carrying Met-Allele
We evaluated the miR-142-3p-associated clinical manifestations in pwRRMS, stratified based on BDNF Val66Met variants.
In Val/Val condition (Table 2), CSF miR-142-3p levels at diagnosis exhibited direct correlation with EDSS (Figure 4A: Spearman’s Rho = 0.326, p = 0.006; n = 69 69), MSSS (Figure 4B: Spearman’s Rho 0.279, p = 0.020, n = 69 69), ARMSS (Figure 4C: Spearman’s Rho 0.287, p = 0.017, n = 69 69) and PI (Figure 4D: Spearman’s Rho = 0.275, p = 0.022; n = 69 69), consisted with the preserved IL1β-miR-142-3p axis (Figure 3A).
Upon dividing patients according to CSF miR-142-3p levels (cut-off: Low miR < 0.01, n = 69 36; High miR ≥ 0.01, n = 69 33), we observed that patients with higher miRNA, showed significantly higher indexes of both disease severity (Figure 4E–G; EDSS: high miR group, median [IQR]: = 2 [1–3] vs. low miR group, median [IQR] = 1.5 [0–2], p = 0.013; MSSS: high miR group, median [IQR]: = 5.9 [1.7–7.4] vs. low miR group, median [IQR] = 2.4 [0.7–4.9], p = 0.022; ARMSS: high miR group, median [IQR]: = 4.1 [2.5–7.3] vs. low miR group, median [IQR] = 3 [1.2–4.7], p = 0.026) and disease progression (Figure 4H; PI high miR group, median [IQR]: = 0.4 [0.0.2–2] vs. low miR group, median [IQR] = 0.04 [0–0.7], p = 0.011) compared to the patients with lower miR-142-3p levels.
Importantly, miR-142-3p was not correlated with any disease parameters in Met-carriers (Table 3; Figure 5A–D: EDSS: p = 0.524; MSSS: p = 0.894; ARMSS: p = 0.936; PI: p = 0.701). No differences emerged even upon patients’ stratification according to miR-142-3p levels (Figure 5E–H; EDSS, p = 0.716; PI, p = 0.641; MSSS, p = 0.973; ARMSS, p = 0.794).
3. Discussion
This study explored the role of CSF miR-142-3p as a biomarker for MS course, particularly in relation to the BDNF Val66Met polymorphism. Our focus was on the early phases of the disease, analyzing pwRRMS at the time diagnosis because the identification of valuable prognostic factors at this stage is urgent for guiding therapeutic choices in the expanding treatment repertoire.
We previously demonstrated in a heterogenous cohort of patients with MS that miR-142-3p is a key regulator of synaptopathy-driven disease progression, which acts as molecular effector of excitotoxic synaptic alterations induced by IL-1β [5,6].
Here, we discovered that the CSF miR-142-3p levels were directly correlated with the different aspects of the RRMS course by evaluating various clinical parameters. EDSS scoring [24] allowed us to assess the disability status of patients. To account for age-related factors, EDSS corrections using ARMSS was applied, as proposed by Alrouji and colleagues [25]. Additionally, MSSS provided indications on the disease disability normalized to disease duration [26]. The rate of disability progression was further evaluated using PI [27].
The assessment of the interplay between genetic variants and an excitotoxic miRNA in determining the clinical manifestations of RRMS represents the primary novelty of this work. Interestingly, we showed that the severity and progression of multiple sclerosis (MS) were not influenced by the CSF levels of miR-142-3p in patients with BDNF Met66-allele background, whereas Val/Val patients with high levels of miR-142-3p exhibited a more severe phenotype.
BDNF Val66Met polymorphism has been described to exert a dichotomous role in MS. Some studies showed a protective role of this polymorphism on the risk of progression in the later stages of the disease [28,29]. Other evidence sustained a detrimental role in early MS stages, with higher radiological activity, lower gray-matter volumes, and higher concentrations of a specific subset of CSF proinflammatory cytokines [21,22]. In our study, we observed that Met-allele, but not the Val/Val genotype, uncoupled the levels of miR-142-3p and IL-1β in the CSF of pwRRMS even though the values were not influenced by the genetic background. This was consistent with the loss of miR-142-3p-dependent disease severity and progression. The molecular underpinnings of the Met-allele epistatic effect on the IL-1β-miR-142-3p axis and on its detrimental action on disease course need to be further investigated. However, this effect could be possibly attributed to the role of both BDNF and miRNA in synaptic transmission.
It is well known from the literature that BDNF Val66Met is involved in an altered intracellular trafficking and secretion of mature BDNF protein in synaptic cleft, impairing synaptic plasticity [19,30]. It cannot be excluded that the Met-allele also influences the ability of BDNF to limit the excitotoxic effects of neuroinflammation when secreted from immune cells in RRMS patients in response to relapses and during remitting phases [31].
A variety of cells expresses miR-142-3p, moving from peripheral monocytes, neutrophils and lymphocytes [32,33,34,35] to infiltrating immune cells, microglia and astroglia [5,8,10]. Increasing evidence in various physiological and pathological conditions suggests the presence of miR-142-3p in neurons [7,36,37,38], likely originating from extracellular vesicles produced by immune cells [35,39].
Many target mRNAs of miR-142-3p participate in immune system-related pathways, such as lymphocyte differentiation, proliferation, and activation [40,41,42,43]. miR-142-3p regulates the post-transcriptional expression of multiple genes involved in kinase signaling (JAK-STAT, MAPK, and PKA pathways) in both immune and nervous systems [10,43,44].
Among other pathways, the dopamine pathway is also affected by miR-142-3p through the direct inhibition of Dopamine Receptor D1 (Drd1) mRNA [45], with a possible implication for nitric oxide release from microglia and brain functioning [45,46].
In recent years, we demonstrated that miR-142-3p can downregulate the protein translation of the glial glutamate–aspartate transporter (GLAST), causing a dysregulation in glutamatergic transmission and consequent excitotoxic synaptic damage in IL-1β-dependent manner [5]. Part of the synaptic action of miR-142-3p might be also mediated by inhibiting microglial BDNF, which acts downstream in the miRNA signal pathway [10]. Although miR-142-3p does not directly bind Bdnf mRNA [15], it mediates Bdnf downregulation by repressing Camk2a, and, thus, the phosphorylation of Cyclic AMP-responsive element-binding protein (CREB), a crucial transcriptional activator of Bdnf expression [10]. It cannot be excluded that this pathway acts in other immune and non-immune cells than microglia, including neurons [7,36,37,38].
The interplay between miR-142-3p and BDNF, with the former acting upstream of the latter, suggests why the synaptotoxic effects of miR-142-3p are fully manifested only in Val/Val genetic context, namely when the BDNF protein can be properly produced and directed to synapses. Conversely, in the presence of Val66Met mutation, pro-BDNF fails to be matured and secreted adequately, thereby disguising the miR-142-3p synaptotoxic effects of miR-142-3p on MS course which are likely, at least in part, dependent on BDNF inhibition. Future studies are needed in this sense.
To the best of our knowledge, our study represents the first exploration in this field and aligns seamlessly with a clinical landscape increasingly oriented toward precision medicine. A primary limitation of our research is the relatively small sample size of enrolled pwRRMS (n = 69 114). A second limitation of our cohort is that the median age at diagnosis of pwRRMS was significantly higher in Met-allele carriers than in Val/Val homozygotes. There was a similar difference, but not significant, in the disease duration. The age of onset of the disease was similar in both groups. Recently, it has been reported improved resilience of neurological functioning to brain aging has been observed in healthy individuals that carrying the Met-allele [47]. This aspect, translated to MS, might explain a potential delay in disease onset due to compensation from clinical symptoms. Future studies are needed to confirm this hypothesis.
Nevertheless, together with further investigations, it may contribute to laying the groundwork for a tailored therapeutic approach based on specific prognostic and predictive biomarkers, including the CSF levels of miR-142-3p and the Val66Met genetic variant. Furthermore, considering the reported modulation of miR-142-3p expression by MS disease-modifying therapies [6,32,48] and its association with treatment response [6], our findings may provide valuable insights to inform the development of personalized treatment strategies for managing MS.
4. Materials and Methods
4.1. Patients with RRMS (pwRRMS)
A cohort of 114 pwRRMS was recruited at Neurology Unity of Neuromed Research Institute in Pozzilli, Italy, to participate in this retrospective observational study (Table 1). All the patients were diagnosed on the basis of clinical, MRI and laboratory evidence, according to 2017 revision of the McDonald criteria [1]. The Ethics Committee of Neuromed Research Institute approved the study (CE 26 October 2017; NCT03217396) according to the Declaration of Helsinki. All patients gave written informed consent to participate in the study. At the time of diagnosis, clinical evaluation and brain and spine Magnetic Resonance Imaging (MRI) were performed. Age, sex, EEDSS, the MSSS, the ARMSS, the PI, the presence of radiological disease activity, and disease duration, measured as the interval between disease onset and diagnosis were included in the evaluation. MRI scan (1.5T) included the following sequences: dual-echo proton density, fluid-attenuated inversion recovery (FLAIR), T1-weighted spin-echo (SE), T2-weighted fast SE, and contrast-enhanced T1-weighted SE before and after intravenous gadolinium (Gd) infusion (0.2 mL/kg). Presence of Gd-enhancing (Gd^+^) lesions defined radiological activity at the time of diagnosis.
4.2. CSF Collection and Analysis
CSF was collected at the time of diagnosis, during hospitalization, by lumbar puncture (LP) for medical purpose. No corticosteroids or disease modifying therapies were administered before LP. Cell-free CSF samples were stored at −80 °C and later analyzed [6]. Briefly, the CSF miR-142-3p levels were detected by quantitative real-time PCR (QIAGEN, Hilden, Germany) and normalized to miR-204-5p [6]. Samples were analyzed in duplicate. CSF concentrations of IL-1β were determined by using a Bio-Plex multiplex cytokine assay (Bio-Rad Laboratories, Hercules, CA, USA) and expressed in picograms/milliliter (pg/mL) according to the standard curve. Samples were analyzed in triplicate.
4.3. SNP Val66Met Analysis
All enrolled patients underwent genotyping for BDNF SNP Val66Met (rs6265). A blood sample (200 μL) was collected at the time of diagnosis. BDNF region containing Val66Met polymorphism was amplified by polymerase chain reaction with the TaqMan method performed using the ABI-Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA; sense primer: 5′-AACCTT-GACCCTGCAGAATG-3′; antisense primer: 5′-ATGGGATTGCACTTGGTCTC-3′) [17]. Sequencing analysis was performed by 10 ng of PCR products, purified with Agenocourt AMPure PCR Purification kit (Agenocourt Bioscience Corporation, Beverly, MA, USA) in accordance with manufacturer’s instructions, using 0.5 pmoles of the sequence primer (5′-AAACATCCGAGGACAAGGTG-3′) and the ABI PRISM BigDye Terminator v3.1 Ready Reaction Cycle Sequencing Kit (Applied Biosystem, Foster City, CA, USA). The sequencing product was purified in using CleanSEQ dye terminal removal kit (Agencourt Bioscience Corporation, Great Boston Area, New England, MA, USA) and run on the Applied Biosystems 3730 DNA Analyzer Instrument (Applied Biosystem, Foster City, CA, USA).
The allele and genotype frequencies of our cohort were compared to the Caucasian population dataset in NHLBI’s TOPMed-BRAVO database.
The Polymorphism Phenotyping v2 (PolyPhen-2) tool was used to predict the possible impact of Val66Met substitution [49]. The HumVar approach was utilized for distinguishing drastic mutation effects, commonly observed in Mendelian disease. The HumDiv approach was applied to evaluate a milder contribution of the genetic variant to pathogenicity in a polygenic or multifactorial context. Variants with scores from 0.0 to 0.15 are predicted to be benign, while a score higher than 0.15 indicates a potentially damaging genetic variant, especially if it ranges from 0.85 to 1.0.
4.4. Statistical Analysis
Shapiro–Wilk test was used to evaluate the normality distribution of continuous variables. Data were shown as mean (standard deviation, SD) or median (interquartile range, IQR). Spearman’s nonparametric correlation was used to test possible associations between variables. Categorical variables were presented as absolute (n) and relative frequency (%). Difference in continuous variables between the BDNF SNP groups was evaluated using the nonparametric Mann–Whitney test. Box plots were employed to highlight statistically significant differences between groups. To distinguish the pwRRMS with low miR-142-3p (Low miR) and the pwRRMS with high miR-142-3p levels in the CSF (High miR), the cut-off value of 0.010 was used [6]. A p value < 0.05 was considered statistically significant.
All the comparisons were performed using Prism GraphPad 9.0 and IBM SPSS Statistics 17.0. for Windows/Mac (IBM Corp., Armonk, NY, USA).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Thompson A.J. Baranzini S.E. Geurts J. Hemmer B. Ciccarelli O. Multiple sclerosis Lancet 20183911622163610.1016/S 0140-6736(18)30481-129576504 · doi ↗ · pubmed ↗
- 2Schwarz K. Schmitz F. Synapse Dysfunctions in Multiple Sclerosis Int. J. Mol. Sci.202324163910.3390/ijms 2402163936675155 PMC 9862173 · doi ↗ · pubmed ↗
- 3Ma Q. Shams H. Didonna A. Baranzini S.E. Cree B.A.C. Hauser S.L. Henry R.G. Oksenberg J.R. Integration of epigenetic and genetic profiles identifies multiple sclerosis disease-critical cell types and genes Commun. Biol.2023634210.1038/s 42003-023-04713-536997638 PMC 10063586 · doi ↗ · pubmed ↗
- 4Mandolesi G. Gentile A. Musella A. Fresegna D. De Vito F. Bullitta S. Sepman H. Marfia G.A. Centonze D. Synaptopathy connects inflammation and neurodegeneration in multiple sclerosis Nat. Rev. Neurol.20151171172410.1038/nrneurol.2015.22226585978 · doi ↗ · pubmed ↗
- 5Mandolesi G. De Vito F. Musella A. Gentile A. Bullitta S. Fresegna D. Sepman H. Di Sanza C. Haji N. Mori F. mi R-142-3p Is a Key Regulator of IL-1β-Dependent Synaptopathy in Neuroinflammation J. Neurosci.20173754656110.1523/JNEUROSCI.0851-16.201628100738 PMC 6596761 · doi ↗ · pubmed ↗
- 6De Vito F. Musella A. Fresegna D. Rizzo F.R. Gentile A. Bassi M.S. Gilio L. Buttari F. Procaccini C. Colamatteo A. Mi R-142-3p regulates synaptopathy-driven disease progression in multiple sclerosis Neuropathol. Appl. Neurobiol.202248 e 1276510.1111/nan.1276534490928 PMC 9291627 · doi ↗ · pubmed ↗
- 7De Vito F. Balletta S. Caioli S. Musella A. Guadalupi L. Vanni V. Fresegna D. Bassi M.S. Gilio L. Sanna K. Mi R-142-3p is a Critical Modulator of TNF-Mediated Neuronal Toxicity in Multiple Sclerosis Curr. Neuropharmacol.2023212567258210.2174/1570159 X 2166623040410391437021418 PMC 10616916 · doi ↗ · pubmed ↗
- 8Junker A. Krumbholz M. Eisele S. Mohan H. Augstein F. Bittner R. Lassmann H. Wekerle H. Hohlfeld R. Meinl E. Micro RNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD 47Brain 20091323342335210.1093/brain/awp 30019952055 · doi ↗ · pubmed ↗