Integrated Analysis of Transcriptome and Metabolome Reveals Differential Responses to Alternaria brassicicola Infection in Cabbage (Brassica oleracea var. capitata)
Jinzhou Lei, Wei Zhang, Fangwei Yu, Meng Ni, Zhigang Liu, Cheng Wang, Jianbin Li, Jianghua Song, Shenyun Wang

TL;DR
This study combines gene and metabolite analysis to understand how cabbage plants resist or succumb to a fungal infection.
Contribution
The study reveals distinct gene and metabolite responses in resistant and susceptible cabbage genotypes to Alternaria brassicicola infection.
Findings
Resistant genotype Bo257 showed fewer differentially expressed genes compared to susceptible Bo190.
Extracellular ROS production genes were largely unchanged in resistant plants but upregulated in susceptible ones.
Succinate accumulation was observed in both genotypes, potentially supporting resistance via energy production.
Abstract
Black spot, caused by Alternaria brassicicola (Ab), poses a serious threat to crucifer production, and knowledge of how plants respond to Ab infection is essential for black spot management. In the current study, combined transcriptomic and metabolic analysis was employed to investigate the response to Ab infection in two cabbage (Brassica oleracea var. capitata) genotypes, Bo257 (resistant to Ab) and Bo190 (susceptible to Ab). A total of 1100 and 7490 differentially expressed genes were identified in Bo257 (R_mock vs. R_Ab) and Bo190 (S_mock vs. S_Ab), respectively. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed that “metabolic pathways”, “biosynthesis of secondary metabolites”, and “glucosinolate biosynthesis” were the top three enriched KEGG pathways in Bo257, while “metabolic pathways”, “biosynthesis of secondary metabolites”, and “carbon metabolism” were…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1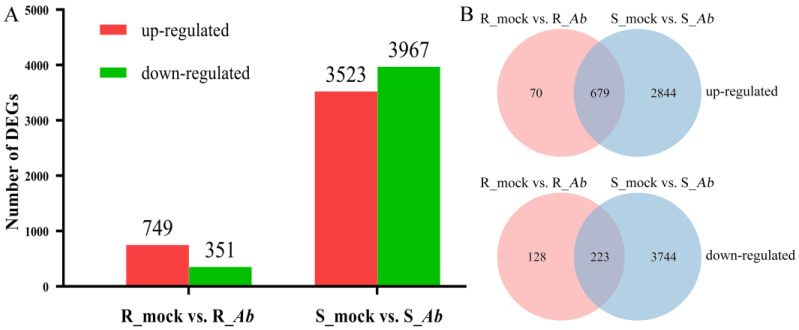 Figure 2
Figure 2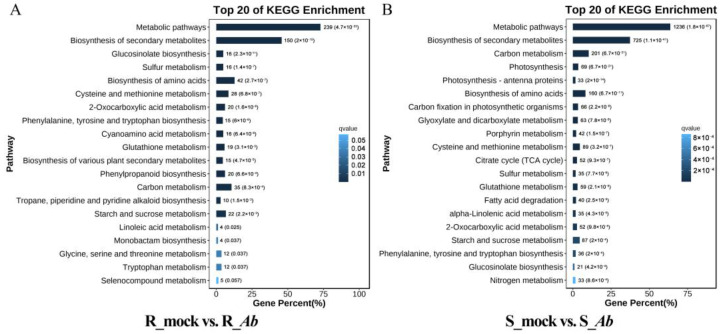 Figure 3
Figure 3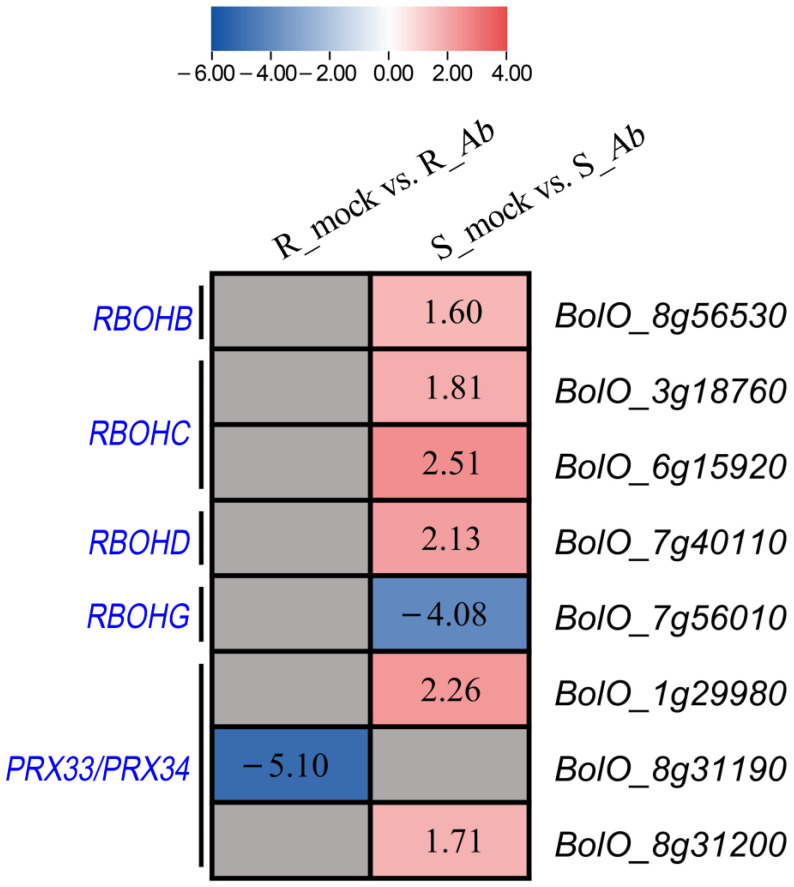 Figure 4
Figure 4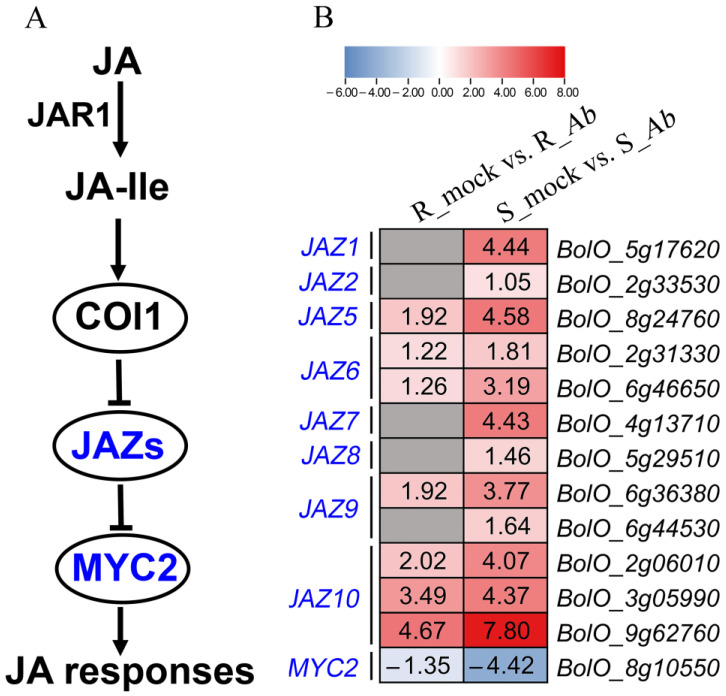 Figure 5
Figure 5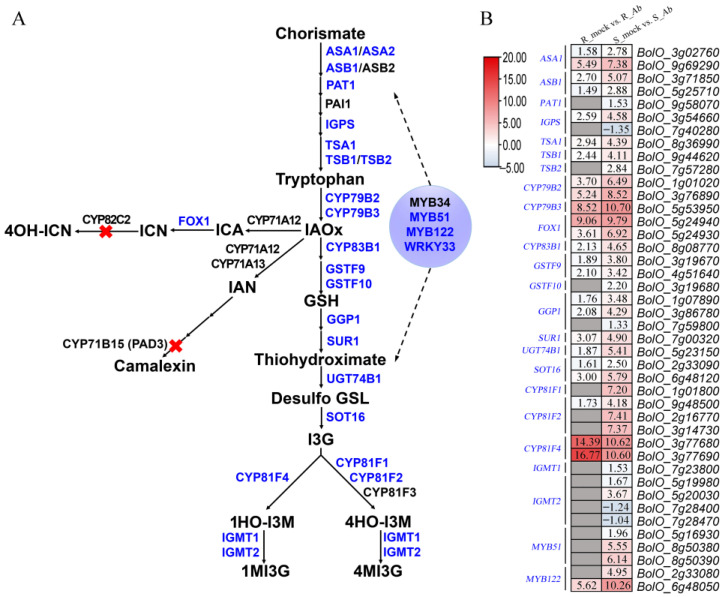 Figure 6
Figure 6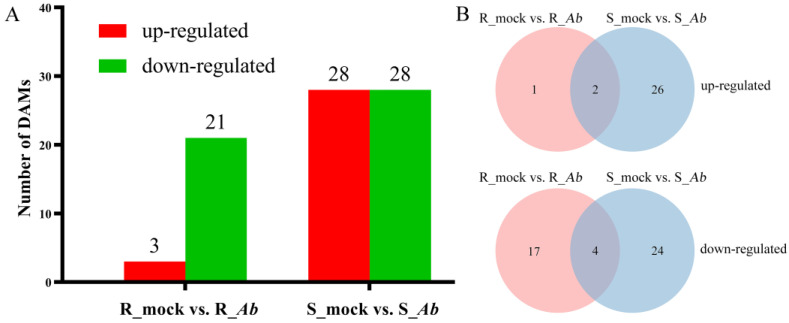 Figure 7
Figure 7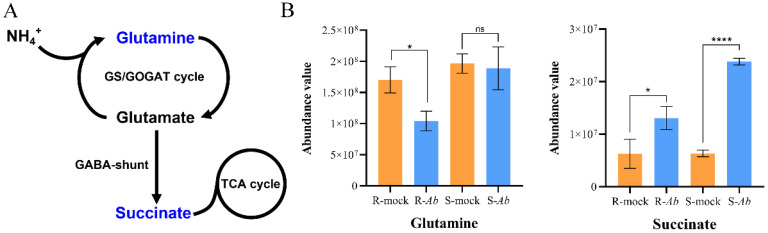 Figure 8
Figure 8- —Key Research and Development Program of Jiangsu Province
- —National Key Research and Development Program of China
- —“JBGS” Project of Seed Industry Revitalization in Jiangsu Province
- —Science and Technology Planning Project of Nanjing City
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Disease Resistance and Genetics · Plant-Microbe Interactions and Immunity · Plant Pathogens and Fungal Diseases
1. Introduction
Alternaria is a large genus mainly consisting of saprophytic fungi; however, this genus also contains some economically important phytopathogens that cause diseases in a wide range of crops like vegetables, fruits, and cereals [1]. Aside from causing yield losses, Alternaria species are known to produce secondary metabolites detrimental for plant growth and human health [2,3]. Among the Alternaria species attacking cruciferous crops, Alternaria brassicae, Ab, Alternaria alternata, and Alternaria raphani reportedly cause substantial damage [4]. In recent years, there has been an increasing incidence of Alternaria black spot (ABS) in the crucifer-growing areas of China, and the development of resistant cultivars is a cost-effective way to control ABS. Unfortunately, the resistant sources imparting complete resistance to ABS are largely unavailable. Only a limited number of resistant sources have been identified in other crucifer species, such as Camelina sativa, Capsella bursa-pastoris, and Arabidopsis thaliana [5,6].
The knowledge of how plants respond to pathogen invasion contributes to the development of strategies for disease management. Rapid production of ROS represents one of the earliest responses upon successful perception of pathogens by plants [7]. The enzymes responsible for extracellular ROS production include cell wall peroxidases, polyamine oxidases, and plasma-membrane-localized NADPH oxidases (respiratory burst oxidase homologs, RBOHs) [8]. The roles of RBOHs and cell wall peroxidases in mediating ABS susceptibility have been documented in A. thaliana [6,9]. Disruption of RBOHD, RBOHE, or RBOHF results in reduced ROS accumulation and cell death, thus conferring various degrees of resistance to A. brassicae [6]. Similarly, knockdown of PEROXIDASE 33/34 (PRX33/PRX34) shows reduced colonization of Ab [9]. Hormone biosynthesis and signaling pathways are also implicated in plant responses to Alternaria infection. A. thaliana mutants defective in different aspects of the auxin pathway are generally more susceptible to Ab as compared with wild-type plants [10]. Disruption of CORONATINE INSENSITIVE 1 (COI1), a gene encoding a critical component of jasmonic acid (JA) signaling, compromises resistance to Ab [11,12]. In addition, abscisic acid, ethylene, and salicylic acid signals have also been implicated in plant responses to ABS [13,14]. Plants also produce multiple secondary metabolites to counteract pathogen invasion. For example, the tryptophan-derived defense-related metabolites camalexin and indolic glucosinolates are involved in resistance to ABS [15,16].
To date, our understanding of plant responses to ABS mainly comes from the Arabidopsis–Alternaria pathosystem, presumably due to the availability of genetic and genomic resources in combination with well-developed molecular tools in A. thaliana. Therefore, it is necessary to investigate how other plant species respond to Alternaria infection. While genetic manipulation remains a time-consuming and challenging task in many horticultural plants, integrated transcriptome and metabolome profiling may provide another avenue. Recently, integrated analysis of transcriptome and metabolome has been widely used to identify the gene candidates and metabolites playing vital roles in biotic and abiotic stresses and quality-related traits [17,18,19]. In the current study, Ab-resistant Bo257 and Ab-susceptible Bo190 were selected from 134 cabbage genotypes. The responses of Bo257 and Bo190 to Ab infection were then investigated. The differentially expressed genes (DEGs) and differentially accumulated metabolites (DAMs) between the mock and Ab-inoculated plants were identified using transcriptomics and widely targeted metabolomics. This study provides comprehensive insights into the interaction between Ab and cabbage, aiding in the discovery of targets for breeding Ab-resistant varieties.
2. Materials and Methods
2.1. Plant Materials, Ab Inoculation, and Sample Collection
Two cabbage genotypes, Bo257 (resistant to Ab) and Bo190 (susceptible to Ab), were utilized in this study and were provided by the Institute of Vegetable Crops, Jiangsu Academy of Agricultural Sciences. HB1 was a field isolate of Ab and was kept at 4 °C before use. The two genotypes (R genotype and S genotype) were grown in a growth chamber at 25 °C under a long-day photoperiod (16 h light/8 h dark). For inoculum preparation, HB1 was cultured on potato dextrose agar (PDA) at 25 °C for 9 days. Subsequently, spores were scraped from sterile distilled-water-flooded PDA plates with a sterile spreader. The resulting suspension was adjusted to 1 × 10^5^ spores mL^−1^ before being uniformly sprayed onto the third true leaf. Plants sprayed with sterile distilled water served as mock controls. The inoculated seedlings were maintained at 25 °C under a relative humidity of approximately 100% for 24 h before being sampled for transcriptomic and metabolomic analysis. The Ab-inoculated samples from Bo257 and Bo190, were labeled as R_Ab (R_Ab1, RAb2, and RAb3) and SAb (S_Ab1, SAb2, and SAb_3), respectively, while the mock-inoculated samples from Bo257 and Bo190 were labeled as R_mock (R_mock_1, R_mock_2, and R_mock_3) and S_mock (S_mock_1, S_mock_2, and S_mock_3), respectively. All the 12 samples were immediately frozen in liquid nitrogen and sent to Gene Denovo Biotechnology Co., Ltd. (Guangzhou, China), for transcriptomic and metabolomic analysis.
2.2. Scanning Electron Microscopy of Cabbage Leaves
Freshly harvested leaves from the two genotypes were carefully washed with distilled water to remove surface dust. The leaves were then cut into pieces measuring approximately 1 cm^2^. Subsequently, fixation was carried out using 2.5% glutaraldehyde for a duration of 2 h. The samples were freeze dried, sputter coated with gold, and finally imaged using Zeiss EVO LS10 scanning electron microscopy (Carl Zeiss, Germany).
2.3. RNA Isolation, Library Construction, and Sequencing
Total RNA was extracted from the cabbage leaves using the Trizol reagent (Invitrogen, USA). Following the RNA quantity and quality assessment, the mRNA was enriched with oligo(dT) beads, and the captured mRNA was fragmented and reversely transcribed into cDNA. The resulting 12 cDNA libraries were sequenced on the Illumina NovaSeq 6000 platform. Three biological replicates were conducted for each experiment.
2.4. Differential Expression Analysis of RNA-Seq Data
Quality control was performed for the raw data by filtering out the adapters, poly-N, and low-quality reads with fastp [20]. The rRNA reads were further removed by mapping with Bowtie2 [21], and the remaining clean reads were then mapped to the cabbage (OX-heart) reference genome using HISAT2 with default parameters [22,23]. The expression levels of the mRNA transcripts were quantified as FPKM (fragments per kilobase of transcript per million mapped reads) using the RSEM v1.3.3 [24]. The DEGs between the treatments (mock and Ab inoculation) were identified according to the following criteria: |log_2_(fold change)| > 1 and false discovery rate (FDR) < 0.05 by the DEseq2 package [25]. Subsequently, Gene Ontology (GO) enrichment analysis and KEGG pathway enrichment analysis for DEGs were performed by the clusterProfiler R package [26], in which the top 20 most represented categories were presented. Heatmaps were generated using TBtools v1.098661 [27].
2.5. Widely Targeted Metabolomic Analysis
The leaf samples were finely ground in liquid nitrogen, and then approximately 100 mg of the powder was subjected to extraction for 12 h at 4 °C with 1.0 mL of 70% methanol solution. After centrifugation, the supernatant was collected and filtered as previously described [28]. The metabolites of the 12 samples were analyzed using a liquid chromatography–electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS) system. The acquired metabolomics data were analyzed according to previously reported methods [29]. DAMs between treatments (mock and Ab inoculation) were screened with a t-test p < 0.05 and variable importance in projection (VIP) ≥1.
3. Results
3.1. Distinct Disease Symptoms in Leaves of Bo257 and Bo190 after Ab Infection
Under the same growth conditions, the leaf of Bo190 (S genotype) showed a shiny green color, while that of Bo257 (R genotype) exhibited a pallid green color (Figure 1A). Examination of the leaf surface by scanning electron microscopy showed that more epicuticular wax was observed on the leaves of Bo257 as compared with those of Bo190 (Figure 1A). To verify the differential responses to Ab infection in the two cabbage lines, leaves of Bo257 and Bo190 seedlings were artificially inoculated with Ab. At 24 h post inoculation (hpi), dark spots were clearly visible on the leaf surface of Bo190, while only a few dark spots were observed on that of Bo257 (Figure 1B), suggesting that the development of black spot symptoms is somehow delayed in Bo257.
3.2. Identification of DEGs after Ab Infection
In order to explore the gene regulatory networks associated with resistance to black spot, a total of 12 RNA samples, collected from three biological replicates of mock and Ab-inoculated leaves of Bo257 and Bo190 at 24 hpi, were subjected to RNA-Seq analysis. Approximately 532.7 million raw reads were generated, yielding an average of 44.4 million reads per sample (Table S1). After quality filtering, approximately 530.5 million reads were obtained. After the removal of rRNA reads, the remaining reads (approximately 528.3 million reads) were aligned to the OX-heart genome [22]. The majority of the reads (approximately 474.2 million) could be mapped uniquely to one location within the reference genome, while a small portion of the reads were either mapped multiple times (approximately 16.3 million reads) or unmapped (approximately 37.8 million reads) (Table S1). The majority of the genes of the biological replicates were clustered together, as revealed by principal components analysis and Pearson’s correlation analysis (Figures S1 and S2).
A total of 1100 and 7490 DEGs were identified in Bo257 (R_mock vs. R_Ab) and Bo190 (S_mock vs. S_Ab), respectively (Figure 2A). After Ab infection, 749 DEGs were upregulated and 351 DEGs were downregulated in the R-genotype Bo257 (Figure 2A). Different from Bo257, 3523 DEGs were upregulated and 3967 DEGs were downregulated in the S-genotype Bo190 (Figure 2A). Of all the upregulated DEGs identified, 679 DEGs were shared by Bo257 and Bo190, while 70 and 2844 DEGs were uniquely identified in Bo257 and Bo190, respectively (Figure 2B). As for the downregulated DEGs identified, 223 DEGs were shared by Bo257 and Bo190, while 128 and 3744 DEGs were uniquely identified in Bo257 and Bo190, respectively (Figure 2B).
3.3. Functional Annotation of DEGs
GO term enrichment analysis was performed to classify their gene functions according to three main GO terms: biological process (BP), cellular component (CC), and molecular function (MF). For the BP ontology, the main enriched terms were “cellular process”, “metabolic process”, and “response to stimulus” (Figure S3). To better understand the biological significance of the gene functions of the identified DEGs, these DEGs were mapped to reference canonical pathways in the KEGG database. Among the top 20 enriched KEGG pathways, 11 were shared by Bo257 and Bo190, including “metabolic pathways”, “biosynthesis of secondary metabolites”, “glucosinolate biosynthesis”, “sulfur metabolism”, “biosynthesis of amino acids”, “cysteine and methionine metabolism”, “2-oxocarboxylic acid metabolism”, “phenylalanine, tyrosine and tryptophan biosynthesis”, “glutathione metabolism”, “carbon metabolism”, and “starch and sucrose metabolism” (Figure 3A,B). Nine pathways, including “cyanoamino acid metabolism”, “biosynthesis of various plant secondary metabolites”, “phenylpropanoid biosynthesis”, “tropane, piperidine and pyridine alkaloid biosynthesis”, “linoleic acid metabolism”, “monbactam biosynthesis”, “glycine, serine and threonine metabolism”, “tryptophan metabolism”, and selenocompound metabolism” were enriched in Bo257 (Figure 3A). By contrast, pathways including “photosynthesis”, “photosynthesis-antenna proteins”, “carbon fixation in photosynthetic organisms”, “glyoxylate and dicarboxylate metabolism”, “porphyrin metabolism”, “tricarboxylic acid (TCA) cycle”, “fatty acid degradation”, “α-Linolenic acid metabolism”, and “nitrogen metabolism” were enriched in Bo190 (Figure 3B).
3.4. Differential Expression of Genes Involved in Extracellular ROS Production
Rapid production of ROS represents one of the earliest responses upon the successful perception of pathogens by plants [7]; therefore, we first investigated the transcript abundance of genes responsible for extracellular ROS production. A total of 23 homologues were identified in OX-heart genome, with 20 genes encoding cell wall peroxidases and 3 encoding plasma-membrane-localized NADPH oxidases (Table S2). Interestingly, one cabbage homologue (BolO_8g31190) of PRX33/PRX34 was downregulated in Bo257 (Figure 4). By contrast, six cabbage homologues, BolO_8g56530 (homologue of RBOHB), BolO_3g18760, and BolO_6g15920 (homologue of RBOHC), BolO_7g40110 (homologue of RBOHD), BolO_1g29980, and BolO_8g31200 (homologue of PRX33/PRX34) were upregulated, while one gene BolO_7g56010 (homologue of RBOHG) was downregulated in Bo190 (Figure 4).
3.5. Differential Expression of Genes Involved in JA Signaling Pathway
Depending on the distinct strategies adopted by plant pathogenic fungi, they are divided into biotrophs, hemibiotrophs, and necrotrophs. As a necrotrophic fungus, Ab infects host tissue and extracts nutrients from dead host cells. JA signaling represents one of the most studied pathways in terms of plant–necrotrophic fungus interaction and thus was reconstructed based on previous studies [30,31,32] (Figure 5A). Therefore, a total of 34 genes responsible for JA signaling pathway were subjected to an examination of transcript abundance in the current study (Table S2). As shown in Figure 5B, thirteen genes were differentially expressed in Bo190, albeit to varying degrees, while only eight DEGs were identified in Bo257. It is well known that JASMONATE ZIM-DOMAIN (JAZ) proteins are key regulators in the JA signaling pathway and function as transcription repressors of JA-responsive genes [33,34]. Interestingly, 12 genes encoding JAZ protein-encoding were upregulated, including JAZ1 (BolO_5g17620), JAZ2 (BolO_2g33530), JAZ5 (BolO_8g24760), JAZ6 (BolO_2g31330 and BolO_6g46650), JAZ7 (BolO_4g13710), JAZ8 (BolO_5g29510), JAZ9 (BolO_6g36380 and BolO_6g44530), and JAZ10 (BolO_2g06010, BolO_3g05990, and BolO_9g62760), while the expression of MYC2 (BolO_8g10550) was decreased in Bo190 (Figure 5B). The Ab inoculation also resulted in an increased expression of JAZ5, JAZ6, JAZ9, and JAZ10 and a decreased expression of MYC2 in Bo257 but to a lesser degree than those in Bo190 (Figure 5B).
3.6. Differential Expression of Genes Involved in Indolic Glucosinolate Biosynthesis Pathway
Glucosinolates, especially indolic glucosinolates, play an important role in the resistance to Ab infection in Arabidopsis, Chinese kale, and broccoli [15,16]; thus, we were interested to investigate the expression profiles of the 72 genes involved in the biosynthesis pathway of indolic glucosinolates in cabbage (Table S2). First, the indolic glucosinolate biosynthesis pathway was reconstructed based on previous studies [35,36,37] (Figure 6A). A total of 42 DEGs were identified in Bo190, containing 39 upregulated DEGs and 3 downregulated DEGs (Figure 6B). By contrast, 25 DEGs were upregulated in Bo257 after Ab infection (Figure 6B). Overall, the expression of genes involved in the biosynthesis pathway of indolic glucosinolates in Bo257 was either unchanged or lower than that in Bo190. However, BolO_3g77680 and BolO_3g77690, homologues of CYP81F4 responsible for the conversion of indole-3-yl-methyl glucosinolate (I3M) to 1-hydroxyindol-3-ylmethyl glucosinolate (1HO-I3M), showed a higher expression in Bo257 (Figure 6B).
3.7. Identification of DAMs after Ab Infection
In order to explore the metabolic changes in cabbage after Ab infection, metabolic profiles of the R-genotype Bo257 and S-genotype Bo190 were analyzed by the LC-ESI-MS/MS method. After quality control, a total of 1112 metabolites were identified from the tested 12 samples. Among them, a total of 24 and 56 DAMs were identified from Bo257 (3 DAMs were upregulated and 21 DAMs were downregulated) and Bo190 (28 DAMs were upregulated and 28 DAMs were downregulated), respectively (Figure 7A). Of all the upregulated DAMs identified, only 2 DAMs were shared by Bo257 and Bo190, while 1 and 26 DAMs were uniquely identified in Bo257 and Bo190, respectively (Figure 7B). As for the downregulated DAMs identified, only 4 DAMs were shared by Bo257 and Bo190, while 17 and 24 DAMs were uniquely identified in Bo257 and Bo190, respectively (Figure 7B). These six DAMs shared by Bo257 and Bo190 were N-methyl-a-aminoisobutyric acid, succinate, methylmalonate, L-asparagine, leucine, and ureidosuccinic acid (Tables S3 and S4). These 74 DAMs were classified into 13 categories, including 36 amino acids and their derivatives, 3 carbohydrates and their derivatives, 9 organic acids and their derivatives, 2 vitamins, 4 nucleotides and their derivates, 4 lipids, 5 organoheterocyclic compounds, 5 alkaloids and their derivatives, 2 phenolic acids, 1 amine, 1 polyamine, 1 phytohormone, and 1 organosulfur compound (Tables S3 and S4). The abundance of DAMs from nine major categories was analyzed (Figure S4). We found that Bo257 and Bo190 contained more primary metabolites (20 and 44) than the secondary metabolites (4 and 12), respectively (Tables S3 and S4). In addition, 50 unique DAMs (26 upregulated and 24 downregulated) in Bo190 were identified. The glutamate-metabolism-related γ-aminobutyrate (GABA) shunt was reconstructed based on previous studies [38,39] (Figure 8A). Compared with R_mock, the accumulation of glutamine was decreased in R_Ab, while this metabolite was not significantly changed in S_Ab as compared with S_mock. The accumulation of succinate was significantly increased in both R_Ab and S_Ab, albeit to different degrees (Figure 8B).
4. Discussion
Black spot caused by Ab is an economically important disease of cabbage. The knowledge of how other plants besides Arabidopsis respond to Ab infection will be helpful to aid in the control of ABS. In the current study, Ab-resistant Bo257 was selected from 134 cabbage accessions. Compared with the susceptible genotype Bo190, more epicuticular wax was observed on the leaf surface of Bo257 (Figure 1A). Meanwhile, when inoculated with Ab, fewer dark spots were visible on the leaf surface of Bo257 (Figure 1B). The cuticular waxes are mainly composed of hydrophobic compounds, including fatty acids and their derivatives such as alkanes, aldehydes, primary alcohols, alky esters, secondary alcohols, and ketones [40]. The roles of cuticular wax in plant defense have been explored in many pathosystems. Epicuticular wax is positively correlated with resistance to A. brassicae in rapeseed and mustard [41]. However, a different scenario was observed in the interaction between Brassica napus and another necrotrophic fungus, Sclerotinia sclerotiorum, where the total amount of wax was significantly lower in the resistant cultivar compared with that in the susceptible cultivar [42]. Sorghum leaf wax improves the growth of Penicillium but suppresses A. alternata, while sheath wax suppresses Penicillium but does not affect A. alternata [43]. In Arabidopsis, the disruption of the LTPG1 gene, encoding a plasma-membrane-localized lipid transfer protein, leads to alterations in cuticular lipid composition but has no significant impact on total wax and cutin monomer loads [44]. Notably, the mutant shows increased susceptibility to Ab than the wild type [44].
Previous studies have suggested that multiple mechanisms are involved in plant resistance against Alternaria species [16,45]. Therefore, to unveil the possible involvement of other regulatory networks in the interaction between cabbage and Ab, the transcriptional landscape was investigated in the Ab-resistant genotype Bo257 and the Ab-susceptible genotype Bo190 that were exposed to Ab infection. Based on Ab infection cycle studies on Brassica oleracea leaves [46,47] and our results (Figure 1B), inoculated cabbage leaves at 24 hpi were sampled for analysis of their transcriptome and metabolome. Interestingly, when infected with Ab, only 1100 DEGs were detected in Bo257 (R_mock vs. R_Ab), while 7490 DEGs were identified in Bo190 (S_mock vs. S_Ab) (Figure 2). In response to Ab invasion, 3844 DEGs were detected in a resistant broccoli, whereas only 1616 DEGs were detected in a susceptible broccoli [16]. In another study, when infected with Xanthomonas campestris pv. campestris, 624 DEGs were detected at 2 dpi in a resistant cabbage line, while 3040 DEGs were detected at 2 dpi in a susceptible cabbage line [19]. GO term enrichment analysis showed that the main enriched terms were “cellular process”, “metabolic process”, and “response to stimulus” (Figure 3A). Furthermore, analysis of enriched KEGG pathways revealed that “metabolic pathways”, “biosynthesis of secondary metabolites”, and “glucosinolate biosynthesis” were the top three enriched pathways in Ab-infected Bo257, while “metabolic pathways”, “biosynthesis of secondary metabolites”, and “carbon metabolism” were the top three enriched pathways in Ab-infected Bo190 (Figure 3B). An essential part of plant apoplastic defense is the production of ROS by the action of RBOHs and cell wall peroxidases [48,49,50]. Of all the 23 gene homologues of RBOHs and PRX33/PRX34 predicted in cabbage (Table S2), only one homologue (BolO_8g31190) of PRX33/PRX34 was downregulated in Bo257 after Ab infection (Figure 4). By contrast, six genes, including RBOHB (1), RBOHC (2), RBOHD (1), and PRX33/PRX34 (2), were upregulated with only one homologue of RBOHG being downregulated in Bo190 (Figure 4). In Arabidopsis, disruption of RBOHs or PRX33/PRX34 results in various degrees of resistance to A. brassicae or Ab infection [6,9], indicating their negative roles in the Arabidopsis–Alternaria interaction. Therefore, it is reasonable to hypothesize that the accumulation of extracellular ROS is not beneficial for cabbage to combat Ab infection. JA signaling is an important part of plant defense against necrotrophic fungus. Surprisingly, JA signaling was somehow inhibited by Ab infection in both Bo257 and Bo190 because an increased expression of JAZ homologues and a decreased expression of MYC2 homologues were observed in both genotypes, albeit to varying degrees (Figure 5B). Further study is needed to clarify the involvement of JA signaling in the cabbage response to Ab infection. The role of indolic glucosinolates in the resistance to Ab infection has been documented in Arabidopsis, Chinese kale, and broccoli [15,16]. A total of 25 and 39 genes involved in the biosynthesis pathway of indolic glucosinolates were upregulated in response to Ab infection in Bo257 and Bo190, respectively (Figure 6B). Notably, BolO_3g77680 and BolO_3g77690, homologues of CYP81F4 responsible for the conversion of indole-3-yl-methyl glucosinolate (I3M) to 1-hydroxyindol-3-ylmethyl glucosinolate (1HO-I3M), showed a higher expression in Bo257 (Figure 6B).
When confronted with biotic stresses, the host plants also undergo metabolic remodeling. Metabolic profiling revealed 24 and 56 DAMs in Bo257 and Bo190, respectively (Figure 7A). This was in agreement with the transcriptomic analysis that showed that a smaller number of DEGs were detected in Bo257 than that in Bo190 (Figure 2), suggesting that Bo257 is much more stable both transcriptionally and metabolically than Bo190 after Ab infection. It is therefore interesting to further investigate the mechanism that Bo257 employs to maintain homeostasis after Ab infection. A total of 20 and 44 differentially accumulated primary metabolites were detected in Bo257 and Bo190, respectively, while 4 and 12 differentially accumulated secondary metabolites were detected in Bo257 and Bo190, respectively (Tables S3 and S4), suggesting the possible role of primary metabolism in plant response to Ab infection. It has been proposed that the role of primary metabolism during plant–pathogen interactions is to maintain cellular energy requirements in host plant defense responses [51,52]. The GABA shunt pathway is able to convert GABA into succinate, thereby connecting the glutamine synthetase (GS)/glutamine 2-oxoglutarate aminotransferase (GOGAT) cycle to the TCA cycle (Figure 8A). The GABA shunt increases succinate formation via three key enzymes: glutamate decarboxylase, GABA transaminase, and succinic semialdehyde dehydrogenase [53]. In the present study, the accumulation of succinate was significantly upregulated in Bo257 and Bo190 after Ab infection, coinciding with a reduced level of glutamine (Figure 8B). The glutamate-metabolism-related TCA cycle pathway plays a critical anabolic role in plant defense against pathogens because the resistance responses are known as highly energy-demanding processes in plants [38,54,55]; therefore, the replenishment of the TCA cycle with succinate presumably provides energy for defense against Ab infection in cabbage.
5. Conclusions
In this study, we found that Bo257 accumulated more epicuticular wax than Bo190, which may contribute partially to Ab resistance. In addition, our work revealed that genes responsible for extracellular ROS production were not actively expressed, while most genes involved in the biosynthesis pathway of indolic glucosinolates were upregulated during Ab infection. Furthermore, Bo257 was able to minimize the damage caused by Ab infection by maintaining metabolic homeostasis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Thomma B.P.H.J. Alternaria spp.: From general saprophyte to specific parasite Mol. Plant Pathol.2003422523610.1046/j.1364-3703.2003.00173.x 20569383 · doi ↗ · pubmed ↗
- 2Lee H.B. Patriarca A. Magan N. Alternaria in food: Ecophysiology, mycotoxin production and toxicology Mycobiology 2015439310610.5941/MYCO.2015.43.2.9326190916 PMC 4505009 · doi ↗ · pubmed ↗
- 3Meena M. Gupta S.K. Swapnil P. Zehra A. Dubey M.K. Upadhyay R.S. Toxins: Potential virulence factors and genes related to pathogenesis Front. Microbiol.20178145110.3389/fmicb.2017.0145128848500 PMC 5550700 · doi ↗ · pubmed ↗
- 4Nowicki M. Nowakowska M. Niezgoda A. Kozik E. Alternaria black spot of crucifers: Symptoms, importance of disease, and perspectives of resistance breeding Veg. Crops Res. Bull.20127651910.2478/v 10032-012-0001-6 · doi ↗
- 5Conn K.L. Tewari J.P. Dahiya J.S. Resistance to Alternaria brassicae and phytoalexin-elicitation in rapeseed and other crucifers Plant Sci.198856212510.1016/0168-9452(88)90180-X · doi ↗
- 6Mandal S. Rajarammohan S. Kaur J. ROS accumulation and associated cell death mediates susceptibility to Alternaria brassicae in Arabidopsis accessions Physiol. Mol. Plant Pathol.2019107515910.1016/j.pmpp.2019.06.001 · doi ↗
- 7Torres M.A. Jones J.D.G. Dangl J.L. Reactive oxygen species signaling in response to pathogens Plant Physiol.200614137337810.1104/pp.106.07946716760490 PMC 1475467 · doi ↗ · pubmed ↗
- 8Waszczak C. Carmody M. Kangasjärvi J. Reactive oxygen species in plant signaling Annu. Rev. Plant Biol.20186920923610.1146/annurev-arplant-042817-04032229489394 · doi ↗ · pubmed ↗