Comprehensive Genetic Evaluation in Patients with Special Reference to Late-Onset Sensorineural Hearing Loss
Ikuyo Miyanohara, Junichiro Ohori, Minako Tabuchi, Shin-ya Nishio, Masaru Yamashita, Shin-ichi Usami

TL;DR
This study shows that genetic factors contribute to late-onset hearing loss, suggesting genetic testing is valuable even for older patients.
Contribution
The study provides a comprehensive genetic evaluation of late-onset hearing loss, revealing a significant diagnostic yield.
Findings
A diagnostic rate of 31.9% was achieved using next-generation sequencing in 91 hearing loss patients.
Genetic causes were identified in 55.9% of patients with familial history and progressive hearing loss.
The diagnostic rate decreased with age, but genetic factors remained relevant even in late-onset cases.
Abstract
Hearing loss (HL) is a common and multi-complex etiological deficit that can occur at any age and can be caused by genetic variants, aging, toxic drugs, noise, injury, viral infection, and other factors. Recently, a high incidence of genetic etiologies in congenital HL has been reported, and the usefulness of genetic testing has been widely accepted in congenital-onset or early-onset HL. In contrast, there have been few comprehensive reports on the relationship between late-onset HL and genetic causes. In this study, we performed next-generation sequencing analysis for 91 HL patients mainly consisting of late-onset HL patients. As a result, we identified 23 possibly disease-causing variants from 29 probands, affording a diagnostic rate for this study of 31.9%. The highest diagnostic rate was observed in the congenital/early-onset group (42.9%), followed by the juvenile/young adult-onset…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
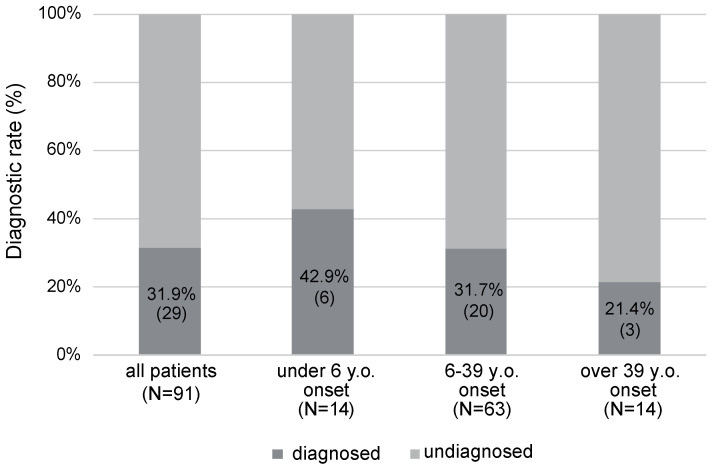 Figure 1
Figure 1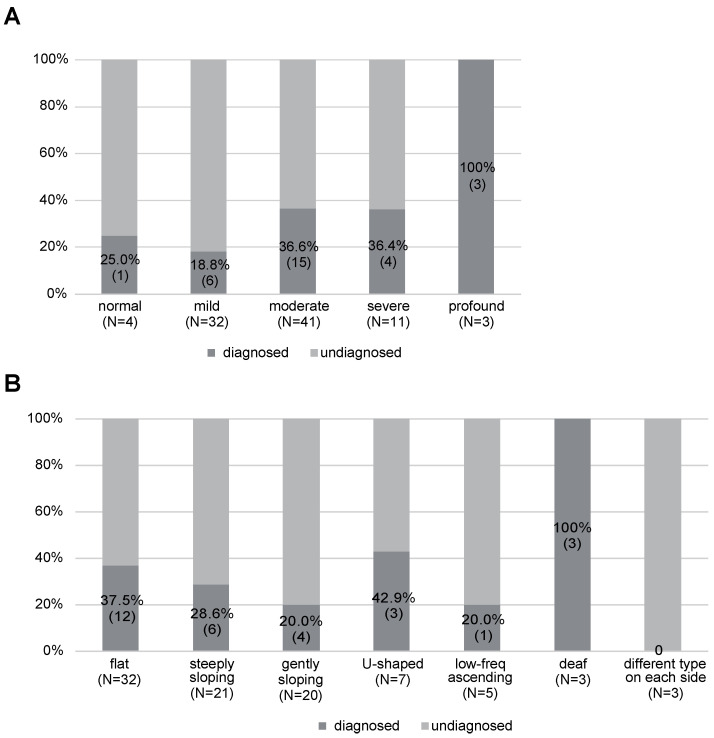 Figure 2
Figure 2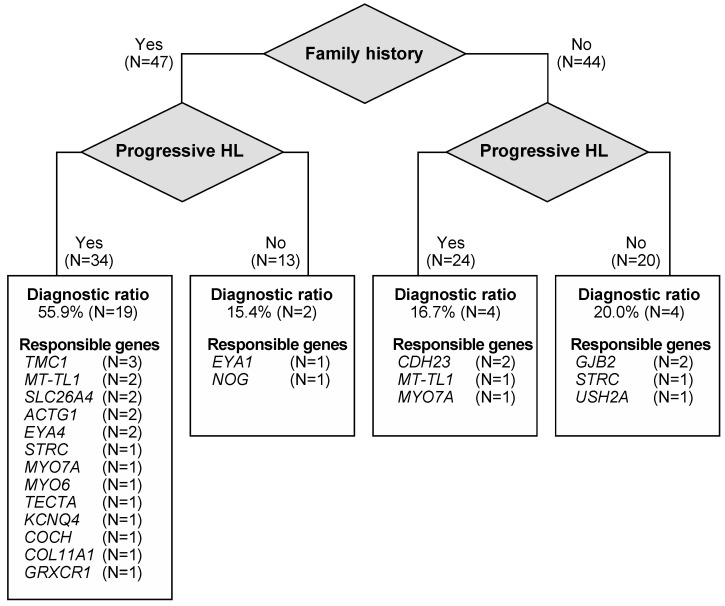 Figure 3
Figure 3- —Health and Labor Sciences Research Grant for Research on Rare and Intractable Diseases and Comprehensive Research on Disability Health and Welfare from the Ministry of Health, Labor and Welfare of Jap
- —Japan Agency for Medical Research and Development (AMED)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHearing, Cochlea, Tinnitus, Genetics · Vestibular and auditory disorders · Ear Surgery and Otitis Media
1. Introduction
Hearing loss (HL) is an etiologically heterogeneous disorder brought about by many causes such as genetic variants, aging, toxic drugs, noise, injury, and viral infections. HL is one of the most common deficits at birth, affecting approximately 1–2 in 1000 newborns [1]. Genetic factors are the most common cause for congenital- or early-onset HL, accounting for at least 60% of congenital sensorineural HL cases [1]. Among these genetic HL cases, the major form of inheritance is autosomal recessive, which is observed in about 75–80% of patients with non-syndromic sensorineural HL [2]. Several studies have focused on congenital- or early-onset HL patients for whom it is possible to achieve higher diagnostic yield [3,4,5,6,7]. In addition, some studies have focused on congenital- or early-onset severe-to-profound HL, for which it is generally possible to obtain a high genetic diagnostic rate [4,5,6,7]. In terms of the disease-causing mechanism, congenital severe-to-profound HL is well explained by genetic factors as pathogenic variants that occurred in the genes essential for the development of hearing function, leading to congenital severe-to-profound HL.
In contrast, the etiologies of late-onset and mild-to-moderate HL remain unclear. Unlike congenital- or early-onset HL, late-onset HL has been considered to result from a variety of factors including genetic and environmental factors. Late-onset HL occurs once hearing function has developed normally, with genetic or environmental factors damaging the maintenance of the hearing function, leading to the onset of HL. It was previously thought that the involvement of genetic factors is limited in late-onset HL. However, recent studies have indicated that a certain proportion of HL that presents after juvenile-onset HL or young adult-onset HL is also due to genetic causes [8,9,10,11].
Therefore, clarifying the genetic etiology, even in late-onset HL, has become more important than ever due to the clinical benefits in providing accurate diagnosis, prediction of HL severity, estimation of associated symptoms, appropriate treatment options, prevention of HL, and better genetic counseling [12]. Here, we report a consecutive prospective study based on target resequencing analysis for HL patients who visited our hospital between March 2007 and September 2022. Our study cohort mainly consists of juvenile- or young adult-onset mild-to-moderate HL patients. We believe our study results will be useful in furthering our understanding of the genetic background of HL patients in real practical clinical settings without any patient selection. In addition, our results are expected to shed some light on the etiology of juvenile- or young adult-onset mild-to-moderate HL.
2. Materials and Methods
2.1. Subjects
Ninety-four patients with HL who visited Kagoshima University hospital between March 2007 and September 2022 were enrolled in this study. We excluded three cases with other etiologies, so that 91 patients with HL and 106 relatives eventually participated in this study. Our university hospital is a tertiary referral hospital, and a wide range of patients in terms of age, particularly late-onset and post-lingual HL patients, are referred to our hospital for examination. In contrast, congenital- or early-onset HL patients identified as part of a newborn hearing screening program usually visit another medical center. Thus, our study cohort mainly consists of late-onset and post-lingual HL patients. This study was conducted with the approval of the ethics committees of Kagoshima University Graduate School of Medical and Dental Sciences and Shinshu University School of Medicine (Approval number: 718). Written informed consent was obtained from all patients (or from their next of kin, caretaker, or legal guardian in the cases of minors or children), and all procedures were performed in accordance with the Declaration of Helsinki Ethical Principles.
2.2. Clinical Evaluations
Clinical information was obtained retrospectively from medical records. Hearing thresholds were evaluated using pure-tone audiometry (PTA) and classified by pure-tone average over 500, 1000, 2000, and 4000 Hz. For infants or young children, conditioned orientation response audiometry and/or auditory brainstem response were performed. The severity of HL was classified into mild (20–40 dB HL), moderate (41–70 dB HL), severe (71–95 dB HL), and profound (>95 dB HL). The audiometric configurations were categorized into low-frequency, mid-frequency (U-shaped), high-frequency (gently sloping-type and steeply sloping-type), flat-type, and deaf, as reported previously [13]. With regard to age at onset, all patients were divided into 3 groups by age; the congenital/early-onset group (under 6 years old), the juvenile/young adult-onset group (6–39 years old), and the middle-aged/aged-onset group (over 39 years old).
2.3. Target Resequencing Analysis
DNA samples extracted from peripheral blood or saliva samples were used in this study. Next-generation DNA sequencing was performed for the 63 target genes [14] reported to be causative for non-syndromic hearing loss (Hereditary Hearing loss Homepage; http://hereditaryhearingloss.org/ accessed on 29 March 2024). We also analyzed 36 previously reported genes for syndromic HL if the patients presented with associated symptoms, as described previously [15]. The detailed protocols and DNA sequencing have been described elsewhere [14]. In brief, amplicon libraries were prepared using the Ion AmpliSeq Custom Panel, with the Ion AmpliSeq Library Kit 2.0 (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. After amplicon library preparation, next-generation sequencing was performed with an Ion Proton or S5 sequencer (ThermoFisher Scientific) according to the manufacturer’s protocol. The sequence data were mapped against the reference human genome sequence (build GRCh37/hg19) with the Torrent Mapping Alignment Program (TMAP). The DNA variants were detected with the Torrent Variant Caller plug-in software (ThermoFisher Scientific).
After variant detection, annotation of identified variants was performed with ANNOVAR software [16]. The missense, nonsense, insertion, deletion, and splicing variants were selected from among the identified variants. Copy number variation analysis was also performed for all patients by using read depth data according to the copy number variation detection methods described in our previous report [17]. Variants were further selected as less than 1% of several control population databases including the 1000 genome database [18], the Genome Aggregation Database [19], the 1200 Japanese exome data in Human genetic variation database [20], the 38,000 Japanese genome variation database [21], and the 333 in-house Japanese normal hearing controls.
The pathogenicity of identified variants was analyzed in accordance with the American College of Medical Genetics (ACMG) standards and guidelines [22] with the ClinGen hearing loss clinical domain working group expert specification [23]. Variants were defined as candidate variants if the following criteria were fulfilled; (1) for the variants previously reported as “pathogenic” or “likely pathogenic” without any contradictory evidence, (2) novel variants classified as “pathogenic” or “likely pathogenic”, (3) variants of “uncertain significance” (VUS) identified as the only candidate after the filtering procedure without any candidate variants among the other 62 genes.
We performed Sanger sequencing analysis to validate the identified variants and confirm family segregations according to the manufacturer’s instructions. All PCR and sequencing primers were designed using the web version Primer 3 plus software [24].
3. Results
3.1. Patient Background and Identified Variants
The age at onset for this study cohort ranged from 0 to 64 years. In this study, we divided patients into three age groups (congenital/early-onset group, juvenile/young adult-onset group and middle-aged/aged-onset group). In general, HL onset under 6 years old is called “pre-lingual onset HL”, and this significantly affects language acquisition. We therefore categorized these patients into one group. It is difficult to differentiate the late-onset HL and presbycusis, so, we divided the other patients by the age of HL onset between 6 and 39 y.o. and over 39 y.o. The former group is thought to consist of late-onset HL patients without presbycusis cases, and the latter group is considered to consist of HL patients including presbycusis patients. The number of patients in each age group was as follows: 14 cases (15.4%) in the congenital/early-onset (onset at under 6 years old) group, 63 cases (69.2%) in the juvenile/young adult-onset (onset at 6–39 years old) group, and 14 cases (15.4%) in the middle-aged/aged-onset (onset at over 39 years old) group (Table 1). One of the unique characteristics of our study cohort was that 84.6% of the patients had late-onset HL.
There are 90 cases with sensorineural HL and one case with mixed HL, with 90 cases showing bilateral HL and one case presenting with unilateral HL. As for the severity, moderate HL was the most common, being observed in 41 cases (45.1%), followed by 32 cases with mild HL (35.2%). As for the audiometric configuration, flat-type HL was the most common, being observed in 32 cases (35.2%), followed by 21 cases with steeply sloping-type HL (23.1%), 20 cases with gently sloping-type HL (22.0%), 7 cases with U-shaped-type HL (7.7%), 5 cases with low-frequency type (5.5%), 3 cases diagnosed as deaf (3.3%), and 3 cases with different types of HL in the left and right ears (Table 1).
Based on the results of the next-generation sequencing analysis, we diagnosed 29 probands among the 91 participants, with the diagnostic rate for this study cohort being 31.9% (Figure 1). The diagnostic rate was highest in the congenital/early-onset group (42.9%), followed by the juvenile/young adult-onset group (31.7%), and the middle-aged/aged-onset group (21.4%) (Figure 1).
We identified 23 disease-causing candidate variants, with the most prevalent responsible genes identified in this study being a mitochondrial m.3243A>G variant and the TMC1 gene, which were observed in 3 cases each, followed by 2 cases with GJB2, CDH23, SLC26A4, STRC, MYO7A, ACTG1, and EYA4 gene variants, respectively, and 1 case each with KCNQ4, MYO6, TECTA, USH2A, COCH, COL11A1, EYA1, NOG, and GRXCR1 variants (Table 2). Among the 23 identified variants, 21 variants had been reported previously, and 2 variants were novel.
3.2. Clinical Characteristics of Patients with Each Gene Variant
HL associated with the GJB2, SLC26A4, STRC, TECTA, USH2A, NOG, and GRXCR1 genes was observed in patients with HL onset in the first decade. In contrast, all patients with TMC1, m.3243A>G, ACTG1, KCNQ4, COCH, COL11A1, EYA1, and MYO7A variants showed HL onset after the second decade. One case with an ACTG1 variant and two cases with MYO7A variants became aware of their HL at over 40 years of age (Table 2).
Patients with EYA1 variants showed mild hearing loss only in the high-frequency region, and these cases were categorized as normal hearing. HL associated with the MYO6, TECTA, and COCH genes was identified in patients with mild hearing loss. Patients with STRC, MYO7A, KCNQ4, USH2, COL11A1, and NOG gene variants showed moderate HL. In terms of audiometric configurations, STRC, EYA4, TECTA, MYO6, USH2, GRXCR1, EYA1, and KCNQ4 gene variants were observed in patients with flat- or U-shaped-type HL. In contrast, patients with TMC1, SLC26A4, COCH, ACTG1, and COL11A1 gene variants presented high-frequency impaired hearing loss, such as steeply sloping or gently sloping audiogram patterns (Table 2).
3.3. The Relationship between Diagnostic Rate and Phenotype
With regard to the diagnostic ratio for each HL severity, the profound HL group showed the highest ratio; i.e., we could identify the responsible gene variants for all three cases (100%), followed by the moderate HL (36.6%), severe HL (36.4%), normal (25.0%), and mild HL (18.8%) groups (Figure 2A). In terms of the diagnostic rate for each type of HL, the deaf-type showed the highest diagnostic ratio (100%), followed by U-shaped HL (42.9%), flat-type HL (37.5%), steeply sloping-type HL (28.6%), gently sloping-type HL (20.0%), and low-frequency ascending-type HL (20.0%) (Figure 2B). We could not identify any candidate variants for the patients with different types of HL on the right and left side.
In addition to the severity of HL and type of HL, family history and progression of HL also affected the diagnostic ratio. There were 47 patients who have relatives with HL, and 44 patients did not have any affected family members. Among the 47 patients with a family history of HL, 34 patients had progressive HL. Similarly, among the 44 patients without a family history, 24 cases had progressive HL (Figure 3). The diagnostic rate was 55.9% for the 34 patients with a family history and progressive HL, which was significantly higher than that for the other groups (p < 0.01, Chi-square test) (Figure 3). Interestingly, most of the responsible genes identified in patients with a family history and progressive HL were autosomal dominant inheritance or maternal inheritance genes, demonstrating the importance of these genes in the patients included in this category.
4. Discussion
In this study, we analyzed 91 HL patients and identified 23 possibly disease-causing variants in 29 cases (31.9%). The diagnostic rate was the highest in the congenital/early-onset group (42.9%), followed by the juvenile/young adult-onset group (31.7%), and the middle-aged/aged-onset group (21.4%). A negative correlation was also observed between the age of onset (or awareness) and diagnostic yield in previous papers [8,9,10,11,45]. These findings emphasize that a genetic etiology is involved to a considerable degree even in patients with late-onset HL.
Previous etiological studies have shown that responsible genes such as GJB2 and SLC26A4 are most frequently identified [4,6]. This result reflects the composition of the study cohort with a particular focus on congenital- or pre-lingual-onset severe-to-profound HL. In this study, most of the participants suffered late-onset mild-to-moderate HL, and we are able to shed some light onto the genetic etiology of late-onset HL patients. The most prevalent responsible genes identified in this study were a mitochondrial m.3243A>G variant and the TMC1 gene, which were observed in three cases each, followed by two cases with GJB2, CDH23, SLC26A4, STRC, MYO7A, ACTG1, and EYA4 gene variants, respectively. The responsible genes identified in this study cohort were consistent with those in our previous report. Usami et al. reported that the frequent responsible genes for juvenile-onset HL were KCNQ4, Mitochondria m.3243A>G variant and m.1555A>G variant, CHD23, MYO6, MYO7A, ACTG1, POU4F3, and WFS1 [9]. One of the major reasons for the large number of late-onset HL patients in this study is that genetic testing for HL has been covered by the social health insurance system in Japan since 2012. Thus, patients from a wide range of age groups and various levels of HL severity can consult specialists for genetic evaluation.
With regard to the relationship between the severity of HL and the diagnostic ratio, a positive correlation was observed. This result was consistent with that of a previous report [9]. In terms of the type of HL, the diagnostic rate was higher for U-shaped HL, flat-type HL, and deaf-type HL than for the other types, including high-frequency steeply sloping-type or gently sloping-type. It might be difficult to distinguish genetic HL from environmental HL in patients with high-frequency-associated HL. Age-related HL and cisplatin-induced HL generally lead to high-frequency HL. Thus, the high-frequency HL group will consist of patients other than those with genetic HL, including those with etiologies such as age, noise, and drugs as well as other environmental factors. Previously, the specific phenotype of a ski-slope audiogram patten, which is one type of high-frequency-associated HL, was also reported to result in a lower diagnostic rate [46]. In addition, both genetic and environmental factors may affect such HL. CDH23 variants cause a wide range of phenotypes, from non-syndromic hearing loss (DFNB12) to syndromic hearing loss and Usher syndrome type ID (USH1D). In addition, several studies proposed that CDH23 variants might modify the susceptibility to HL caused by the environmental factors such as age-related changes or noise exposure [47,48,49,50,51,52]. Thus, CDH23 variants that are observed at high frequencies in the normal hearing control population may affect late-onset high-frequency HL in combination with environmental factors.
Interestingly, family history and progressive HL appear to be good markers for a higher diagnostic ratio. In this study, a higher diagnostic ratio was achieved in 19 (55.9%) of 34 patients with both a family history and progressive HL, even though most of these cases had late-onset mild-to-moderate HL. Among these 19 cases, 13 cases were diagnosed with autosomal dominant inheritance gene-associated HL, and 3 cases were diagnosed with maternal inheritance. In general, autosomal dominant non-syndromic HL is considered to be associated with post-lingual-onset, progressive HL [12], and our results were consistent with this hypothesis.
While useful findings were obtained, this study is limited by the small number of patients accumulated from the single institute. Further studies with a larger number of patients will be required to clarify the characteristics of each type of genetic HL.
5. Conclusions
In conclusion, we showed the utility of genetic testing even in the cases with late-onset HL. In particular, patients with late-onset mild-to-moderate HL with a family history and progressive HL appear to be good candidates for genetic testing. Our data will be useful in furthering our understanding of the genetic background of late-onset mild-to-moderate HL.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Morton C.C.A. Nance W.E. Newborn hearing screening—A silent revolution N. Engl. J. Med.20063542151216410.1056/NEJ Mra 05070016707752 · doi ↗ · pubmed ↗
- 2Smith R.J. Bale J.F. White K.R. Sensorineural hearing loss in children Lancet 200536587989010.1016/S 0140-6736(05)71047-315752533 · doi ↗ · pubmed ↗
- 3Bademci G. Foster J.2nd Mahdieh N. Bonyadi M. Duman D. Cengiz F.B. Menendez I. Diaz-Horta O. Shirkavand A. Zeinali S. Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort Genet. Med.20161836437110.1038/gim.2015.8926226137 PMC 4733433 · doi ↗ · pubmed ↗
- 4Yang T. Wei X. Chai Y. Li L. Wu H. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing Orphanet J. Rare Dis.201388510.1186/1750-1172-8-8523767834 PMC 3703291 · doi ↗ · pubmed ↗
- 5Walsh T. Abu Rayan A. Abu Sa’ed J. Shahin H. Shepshelovich J. Lee M.K. Hirschberg K. Tekin M. Salhab W. Avraham K.B. Genomic analysis of a heterogeneous Mendelian phenotype: Multiple novel alleles for inherited hearing loss in the Palestinian population Hum. Genom.2006220321110.1186/1479-7364-2-4-203PMC 352515216460646 · doi ↗ · pubmed ↗
- 6Yuan Y. Li Q. Su Y. Gao X. Liu H. Huang S. Kang D. Todd N.W. Mattox D. Zhang J. Comprehensive genetic testing of Chinese SNHL patients and variants interpretation using ACMG guidelines and ethnically matched normal controls Eur. J. Hum. Genet.20202823124310.1038/s 41431-019-0510-631541171 PMC 6974605 · doi ↗ · pubmed ↗
- 7Ma J. Ma X. Lin K. Huang R. Bi X. Ming C. Li L. Li X. Li G. Zhao L. Genetic screening of a Chinese cohort of children with hearing loss using a next-generation sequencing panel Hum. Genom.202317110.1186/s 40246-022-00449-1PMC 981174536597107 · doi ↗ · pubmed ↗
- 8Sloan-Heggen C.M. Bierer A.O. Shearer A.E. Kolbe D.L. Nishimura C.J. Frees K.L. Ephraim S.S. Shibata S.B. Booth K.T. Campbell C.A. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss Hum. Genom.201613544145010.1007/s 00439-016-1648-826969326 PMC 4796320 · doi ↗ · pubmed ↗