NF1 with 47,XYY mosaicism diagnosed by mandibular neurofibromas
Erina Tonouchi, Kei-ichi Morita, Yosuke Harazono, Kyoko Hoshino, Tetsuya Yoda

TL;DR
A patient with Neurofibromatosis type 1 and 47,XYY mosaicism was diagnosed through skin and jaw tumors.
Contribution
A novel pathogenic NF1 mutation was identified in a patient with 47,XYY mosaicism.
Findings
Café-au-lait macules and mandibular neurofibromas led to the diagnosis of NF1.
A novel frameshift mutation in the NF1 gene was detected via targeted sequencing.
The patient also exhibited 47,XYY mosaicism, a rare chromosomal abnormality.
Abstract
Neurofibromatosis type 1 (NF1) is an autosomal dominant nevus disease characterized by multiple manifestations, primarily café-au-lait macules and neurofibromas. Here, we present the case of an NF1 patient with 47,XYY mosaicism whose diagnosis was prompted by café-au-lait macules on the skin and mandibular neurofibromas. Targeted next-generation sequencing of the patient’s blood sample revealed a novel frameshift mutation in NF1 (NM_000267.3:c.6832dupA:p.Thr2278Asnfs*8) that is considered a pathogenic variant.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
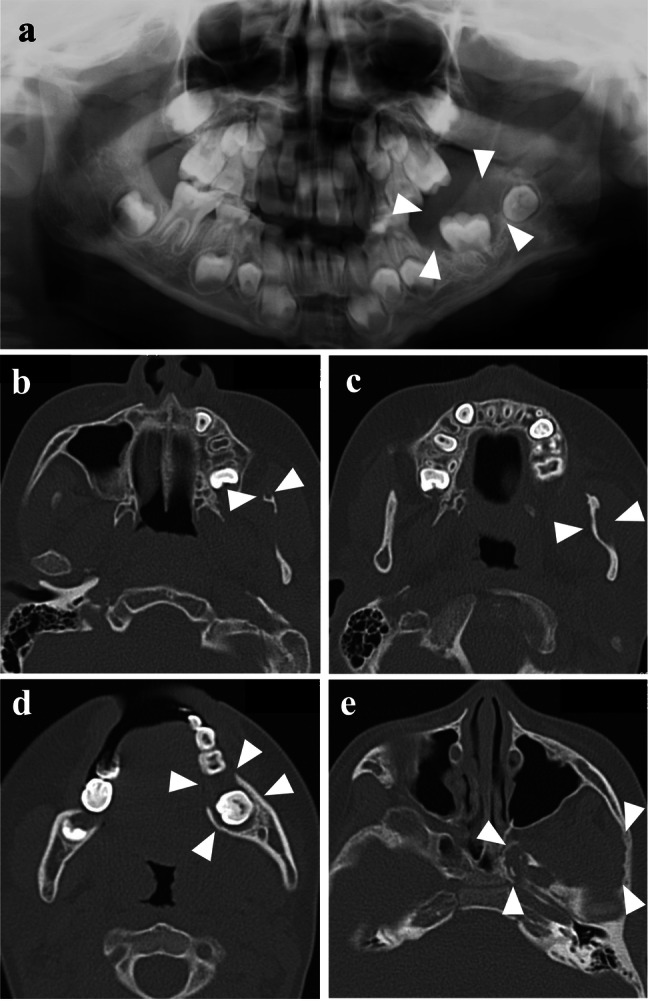 Figure 1
Figure 1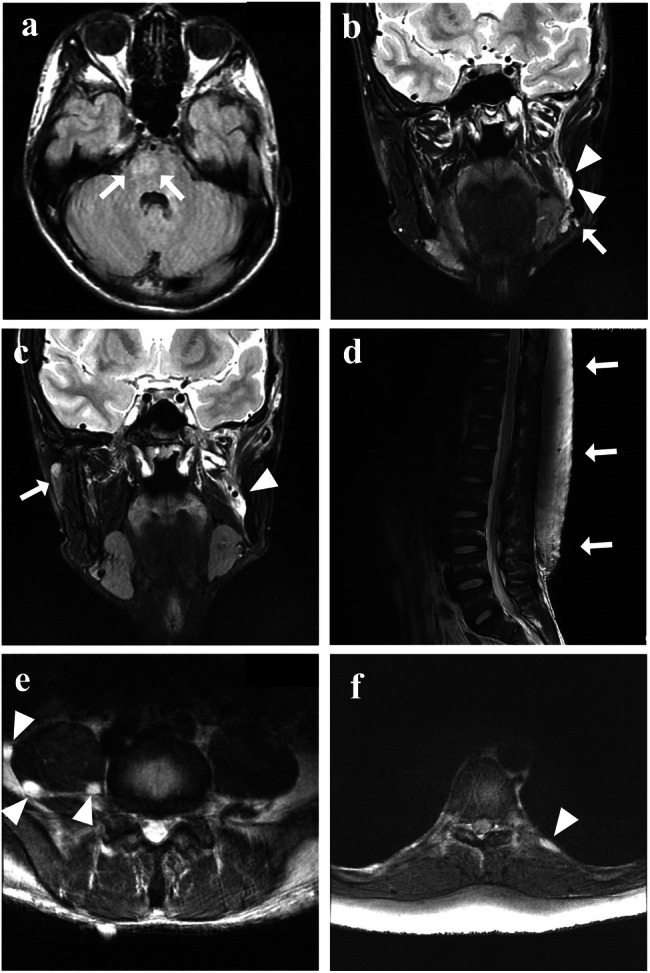 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeurofibromatosis and Schwannoma Cases · Sarcoma Diagnosis and Treatment · Meningioma and schwannoma management
Neurofibromatosis type 1 (NF1; OMIM 162200), an autosomal dominant nevus disease with multiple manifestations, mainly café-au-lait macules and neurofibromas, affects 1 in 3000 people, and approximately 50% of cases arise de novo^1^. The causative gene, NF1 (17q11.2; NM_001042492.3), was identified in 1990^2–4^ and encodes the GTPase-activating protein (GAP) neurofibromin^5^. The diagnostic criteria for NF1 were outlined at the National Institutes of Health (NIH) Consensus Development Conference in 1988^6^ and revised in 2021^7^. Approximately 7% of NF1-related neurofibromas manifest in the oral cavity, and mandibular occurrence is rare^8^. The incidence of 47,XYY syndrome in live-born male infants is 1 in 1000^9^, and the disorder is commonly diagnosed in the first decade of life based on developmental delays, behavioral issues, and tall stature^10^. Herein, we report a novel NF1 frameshift mutation in a patient with NF1 and 47,XXY/46,XY mosaicism whose NF1 diagnosis was prompted by mandibular neurofibromas.
At 7 years of age, the patient visited Tokyo Medical and Dental University Hospital’s oral surgery department following a radiolucent finding around a buried left mandibular first molar on radiographic examination. His medical history included epilepsy, intellectual disability, autism spectrum disorder, and a mosaic karyotype of 47,XYY (43.3%)/46,XY (56.7%), identified through chromosome karyotyping at age 1. The patient’s parents showed no abnormalities, and the family history was unremarkable. The left mandibular first molar had not erupted, and panoramic radiography revealed a radiolucent area around its crown (Fig. 1a). A biopsy confirmed the presence of neurofibroma around the left mandibular first molar. Skin examination revealed multiple café-au-lait macules throughout the body. Clinical diagnosis of NF1 was based on these macules and multiple neurofibromas observed in the left mandibular coronoid process, left mandibular ramus, left mandibular body (biopsy area), and adipose tissue of the left masticator space on computed tomography (CT) scans at age 9 (Fig. 1b–e). Jaw neurofibromas were managed through observation. However, given that the left mandibular first molar had not erupted, the tumor in that area was removed, and fenestration surgery was performed at 10 years of age. Magnetic resonance imaging (MRI) at 16 years of age revealed a right pontine hamartomatous lesion associated with NF1 (Fig. 2a), a 35 mm neurofibroma in the medial mandibular ramus (Fig. 2b), and suspected neurofibromas in the right buccal (Fig. 2c) and left mandibular subcutaneous (Fig. 2b) regions. MRI at age 22 showed lumbar subcutaneous adenomas (Fig. 2d) and neurofibromas around the bilateral psoas major muscle (Fig. 2e) and along the pleura (Fig. 2f). By age 29, axillary and inguinal freckling was confirmed, prompting genetic testing of a peripheral blood sample. This study was approved by the Ethics Review Committee of our institution (D2020-084), and written informed consent was obtained from the parents of the patient. Targeted next-generation sequencing of NF1, NF2, SPRED1, SMARCB1, and LZTR1 using the hybrid capture method at the Kazusa DNA Research Institute, Chiba, Japan, identified a heterozygous mutation, c.6832dupA (p.Thr2278Asnfs*8), in the NF1 gene (NM_000267.3). Sanger sequencing confirmed that the patient had the same mutation (Supplementary Information 1). This NF1 frameshift variant was not registered in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), gnomAD (https://gnomad.broadinstitute.org/), ToMMo (https://jmorp.megabank.tohoku.ac.jp/), or HGMD (https://www.hgmd.cf.ac.uk/ac/index.php). The mutant was a null variant, absent from the gnomAD database, and assumed to be de novo, corresponding to PVS1, PM2, and PM6 in the American College of Medical Genetics and Genomics criteria, respectively; therefore, this variant was considered pathogenic^11^. With no notable changes in intramandibular or perimandibular lesions or systemic subcutaneous lesions, the patient remained under observation.Fig. 1. Panoramic radiography and CT imaging of the mandible.a Panoramic radiography at the initial examination (age 7). b–e CT imaging revealed multiple neurofibromas at age 9 in the left mandibular coronoid process (b), left mandibular ramus (c), left mandibular body (d), and adipose tissue of the left masticator space (e). Arrowheads indicate neurofibromas.Fig. 2MRI findings.a Brain MRI axial FLAIR image at age 16. Arrows indicate hamartomatous lesions. b, c Coronal T2-weighted brain MR image at age 16. The arrows indicate nodules suspected to be neurofibromas in the left mandibular (b) and right buccal (c) subcutaneous areas. Arrowheads indicate neurofibromas. d Sagittal T2-weighted view of a lumbar MR image at age 22. The arrows indicate lumbar subcutaneous adenomas. e, f Axial T2-weighted views of a lumbar MR image at age 22. The arrows indicate neurofibromas around the bilateral psoas major muscle (e) and along the pleura (f).
Neurofibromatosis induces various abnormalities, particularly neurofibromas in the skin and nerves. In this patient, the NF1 diagnosis arose based on the presence of mandibular neurofibromas and café-au-lait macules on the skin. ~7% of neurofibromas in patients with NF1 are reported in the oral cavity^8^, with the palate, gingiva, tongue, buccal mucosa, and lips being common sites; however, mandibular occurrence is rare^8^. In the present case, despite the presence of café-au-lait macules as an NF1 clinical manifestation, the diagnosis was established only when mandibular neurofibromas were identified. Other developmental delays were noted, potentially attributed to the 47,XYY mosaic. Early recognition of comorbidities and comprehensive clinical assessments, facilitated by a timely diagnosis via a multidisciplinary approach, contribute to improving long-term health prognoses.
The diagnosis of 47,XYY is often based on behavioral and learning disabilities^3^. Despite its prevalence (1 in 1000 boys), ~85% of cases are undiagnosed until infertility arises due to atypical symptoms^9^, and limited information is available regarding the phenotype^10^. In our patient, chromosome karyotyping was performed at age 1 owing to developmental delays; however, the 43.3% mosaic nature of 47,XYY complicates our understanding of its impact on the patient’s clinical manifestations.
NF1, the first human condition attributed to pathogenic variants in RAS pathway genes, falls within the broader group of RASopathies, which includes Noonan, Costello, and cardiofaciocutaneous syndromes^12^. Legius syndrome (OMIM 611431), caused by a heterozygous pathogenic variant of SPRED1, is a RASopathy that exhibits the most overlap with NF1^13^. Because other syndromes exhibit clinical manifestations similar to those of NF1, a simultaneous search for candidate pathogenic genes, including SPRED1, is useful for confirming genetic diagnoses. Although our case involved a relatively easy clinical diagnosis of NF1, simultaneous gene searches are crucial in patients with insufficient clinical manifestations for disease differentiation.
The variable phenotype of NF1, even in patients with identical genetic variants, poses challenges in predicting disease severity, including within families^1,14^. This variability may be attributed to factors such as genetic modifiers, epigenetic abnormalities, and environmental influences^14^. Clinical genetic testing, which has high detection rates and utility in identifying genotype–phenotype correlations^5^, has become crucial. In the present case, the identified frameshift mutation, with a stop codon behind the RAS-GAP domain, was revealed through next-generation sequencing.
Previous studies have aimed to identify genotype–phenotype correlations that can be used to predict disease progression. However, mutation hotspots are lacking, and knowledge of genotypes associated with specific phenotypes is limited^15^. Scala et al. reported that missense mutations were inversely associated with neurofibromas, whereas frameshift mutations and whole-gene deletions were associated with skeletal abnormalities^16^. Gjorgjievska et al. reported positive correlations between cognitive impairment and gross deletions or truncating variants^17^. Napolitano et al. reported increased odds ratios for learning disabilities in patients carrying frameshift mutations^15^.
Symptom appearance varies, with café-au-lait macules and neurofibromas potentially manifesting by age 1, whereas other symptoms may develop in childhood, requiring up to 20 years for a patient to meet all diagnostic criteria. Recent advancements in NF1 genetic testing have provided a clinically available approach with a high detection rate, enabling earlier diagnosis in suspected patients.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at 10.6084/m9.figshare.hgv.3400.
Supplementary information
Supplementary information 1