A patient with albinism and retinitis pigmentosa, a case report
Michalis Georgiou, Shaima Awadh Hashem, Michel Michaelides, Joseph G. Chacko, Sami H. Uwaydat

TL;DR
A patient with albinism and retinitis pigmentosa is reported, highlighting the challenges in diagnosing RP in individuals with albinism.
Contribution
This is the first documented case of a patient with both oculocutaneous albinism and retinitis pigmentosa.
Findings
The patient had a homozygous TYR variant causing albinism and a PDE6A variant causing retinitis pigmentosa.
Fundus autofluorescence imaging showed features consistent with retinitis pigmentosa despite albinism-related hypopigmentation.
Genetic testing confirmed the dual diagnosis, aiding in the identification of an extremely rare condition.
Abstract
To present a case of molecularly confirmed oculocutaneous albinism (OCA) and retinitis pigmentosa (RP). A 46-year-old male with a lifelong established diagnosis of OCA and baseline best corrected visual acuity (BCVA) of 20/200, presented for worsening visual acuity over the last few years. BCVA was light perception and hand motion at face for the right and left eye, respectively. Fundus exam showed hypopigmented fundi with visible choroidal vessels and blunted foveal reflexes in both eyes. Optical coherence tomography showed foveal hypoplasia and outer retinal degenerative changes not typical of OCA. Fundus autofluorescence (FAF) imaging showed focal areas of decreased signal at the fovea, similar to areas of atrophy in an age matched patient with PDE6A-RP. Genetic testing identified a homozygous disease-causing variant in TYR c.1467dup, p. (Ala490Cysfs*20) causing OCA, and a…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
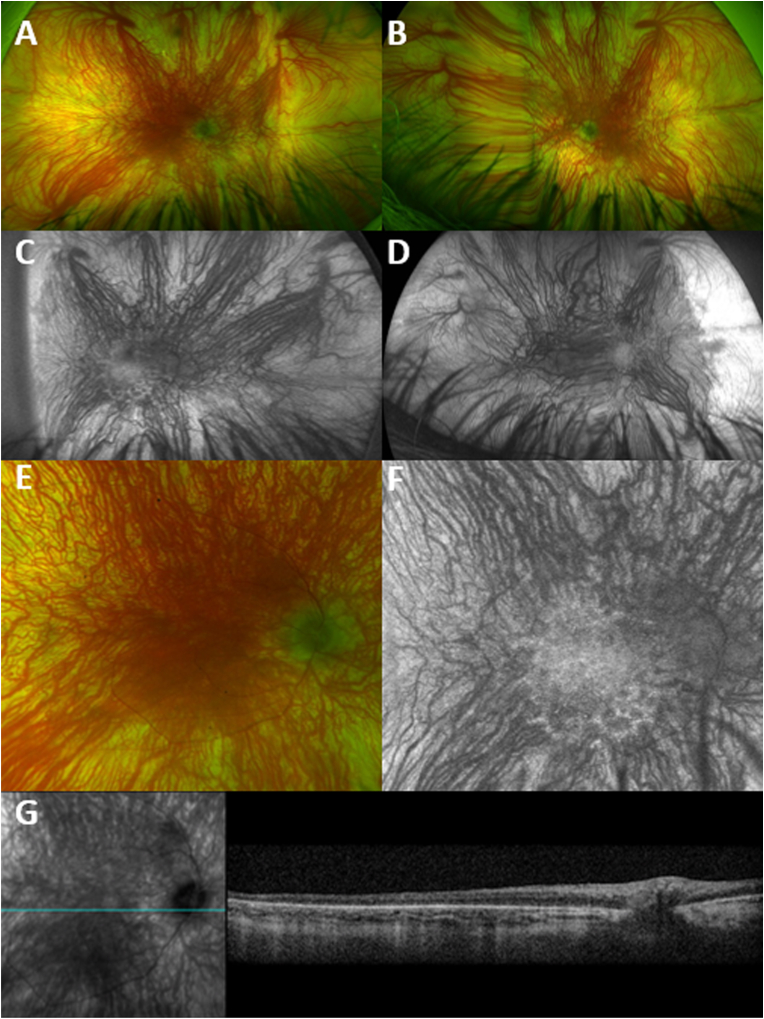 Figure 1
Figure 1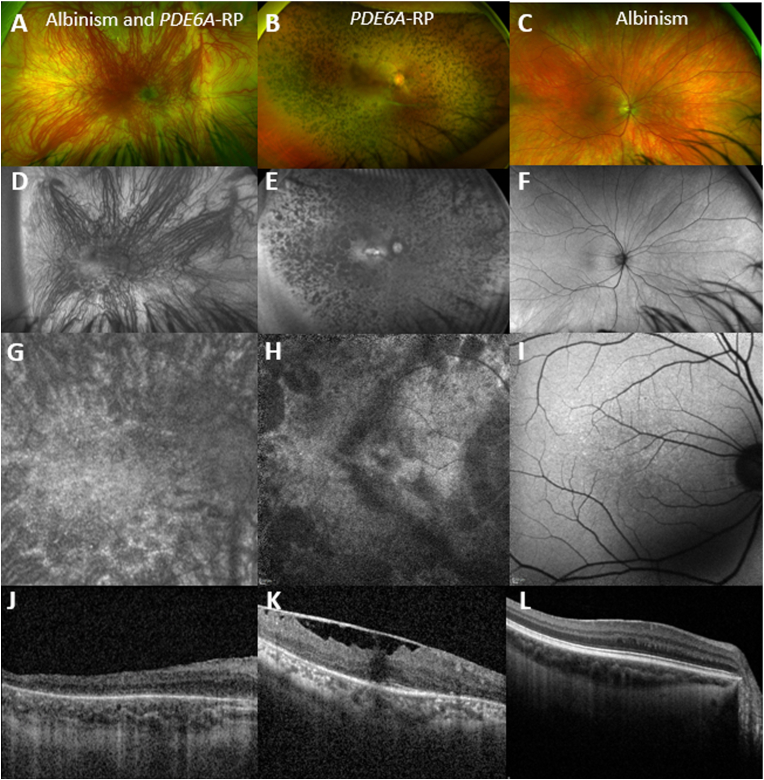 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRetinal Development and Disorders · melanin and skin pigmentation · Retinal Diseases and Treatments
Introduction
1
Retinitis pigmentosa (RP) is the most common inherited retinal degeneration phenotype and is characterized by nyctalopia and gradual constriction of the visual field, with eventual loss of central vision, progressing to legal blindness. RP can be inherited as an autosomal dominant (AD), autosomal recessive (AR) or X-linked (XL) trait.1 Oculocutaneous albinism (OCA) is a group of genetic disorders characterized by general skin, hair and retinal hypopigmentation, most commonly inherited as an AR condition.2 Major and minor criteria for the diagnosis of OCA have been recently proposed. Major criteria include: (1) grade 2 or more foveal hypoplasia, (2) optic nerve misrouting, and (3) ocular hypopigmentation, with either iris translucency or grade 2 or more fundus hypopigmentation. Minor criteria include: (1) nystagmus, (2) hypopigmentation of skin and hair, (3) grade 1 fundus hypopigmentation, and (4) grade 1 foveal hypoplasia. Kruijt et al. proposed that 3 major criteria or 2 major and 2 minor criteria are necessary for the diagnosis OCA. In the presence of a molecular diagnosis, 1 major criterion or 2 minor criteria is considered sufficient.2
Herein we report a molecularly confirmed patient with OCA and RP. We describe in detail the presentation of this rare entity.
Case report
2
A 46-year-old white male with lifelong established clinical diagnosis of OCA, without molecular confirmation, presented to our ocular genetics clinic (Jones Eye Institute, University of Arkansas Medical Science, Little Rock, AR) for worsening vision. Based on the referral records, the patient had a best corrected visual acuity (BCVA) of 20/200 for both eyes for most of his life, and nystagmus since birth. Patient reported progressive vision loss over the last few years, prompting the referral.
Pupils were equal, 4mm and minimally reactive. BCVA was light perception (LP) and hand motion (HM) at face for the right and left eye respectively, not improving with pinhole or refraction. Patient could not identify any of the Ishihara colour plates due to the severely reduced vision. Intraocular pressure was 15/16 mmHg. Confrontational visual fields could not be assessed. Ocular motility was intact, and severe nystagmus was noted. Examination of the anterior segment was remarkable for 360° iris transilluminations and mild posterior subcapsular cataract in both eyes. Dilated fundoscopy (DFE) revealed a blunted foveal reflex, blonde fundi and visible choroidal vessels, with generalised retinal vessel attenuation (Fig. 1A–B). Optic discs had a gray appearance and were sharp with 0.1 cup/disc ratio in both eyes.Fig. 1. Multimodal Macular Imaging of a Patient with Retinitis Pigmentosa and Albinism.Pseudocolor fundus photographs of the right (A) and left (B) eye of a patient with PDE6A-retinitis pigmentosa and TYR-albinism, with bilateral blunted reflex, attenuated vessels, small and gray appearing disks. Fundus autofluorescence (FAF) of the right (C) and left (D) eye of the same patient with profound choroidal vessel show. Spotty areas of decreased signal are visible within the arcades, easier to appreciate at the inferior arcade of the right eye (C), due to better imaging quality, which are not usually present in patients with albinism. (E) Pseudocolor fundus magnified image of the macula. (F) Magnified FAF imaging showing profound choroidal vessel show, and patchy areas of signal loss inside the vascular arcades. (G) Infrared image showing the location of the transfoveal optical coherence tomography (OCT) scan. OCT shows grade 4 foveal hypoplasia, with absence of the normal widening of the outer nuclear layer, a finding common in albinism. In contrast to albinism, the ellipsoid zone is disrupted/attenuated. Retinal imaging was challenging due to nystagmus and associated artefacts.Fig. 1
Retinal imaging
3
Ultra widefield fundus autofluorescence (FAF, Optos Silverstone, Marlborough, MA, USA) imaging was significant for extensive choroidal show, attenuated retinal vessels and decreased signal within the arcades, which typically are not findings in patients with OCA (Fig. 1C–D). Optical coherence tomography (OCT, Spectralis, Heidelberg Engineering Franklin, MA, USA) showed grade 4 foveal hypoplasia, Fig. 1G. Ellipsoid zone (EZ) was disrupted and attenuated, which is not a typical presentation of OCA. Retinal imaging was challenging due to nystagmus.
Genetic testing
4
The patient underwent genetic panel testing, which included ssequence analysis and deletion/duplication testing of 314 genes (Blueprint Genetics, My Retina Tracker Program Panel, version 10, Jul 10, 2023), and was found to be homozygous for disease causing variants (classified as pathogenic) in the TYR and PDE6A genes. Patient was homozygous for the TYR (NM_000372.5): c.1467dup, p. (Ala490Cysfs*20) variant that generates a frameshift resulting in a premature stop codon in the last exon, and for the PDE6A (NM_000440.3): c.304C > A, p. (Arg102Ser) missense variant. There are other published reports of the same phenotype caused by other truncating variants 3′ side in the last exon of the TYR gene (NM_000372.5 (TYR):c.1501dup (p.Arg501fs)). This can support moderate to strong evidence of the pathogenicity based on the ACMG guidelines.3 There are several published reports of the same PDE6A gene variant in multiple families, and the variant is classified as pathogenic.
Controls
5
Given the inherent variability of presentation in patients with inherited retinal diseases, we performed a search on the genetic database of the Jones Eye Institute, University of Arkansas Medical Science, Little Rock, AR, and of the Moorfields Eye Hospital (MEH), London, UK, for patients with isolated PDE6A-RP or TYR-albinism with the same variants and of similar age.
We identified a matching 48 year old female control in the MEH database, homozygous for the same PDE6A variant. Age of disease onset was 12 years old, presenting with nyctalopia. BCVA was 0.3 LogMAR (20/40) and 0.5 LogMAR (20/60). Patient had normally pigmented iris, pupils were equal and reactive, and trace posterior subcapsular changes were present. DFE showed midperipheral bone-spicule-like retinal pigmentary changes and atrophic changes extending from the periphery to the vascular arcades (Fig. 2 B). Vessel attenuation, epiretinal membrane, and waxy disc pallor were also observed. Widefield FAF showed patchy areas of decreased signal in the mid-periphery and up to the vascular arcades (Fig. 2 E). Macular FAF showed similar pattern of signal changes to the patient with OCA and RP (Fig. 2 G and H). OCT showed better preserved foveal EZ compared to the patient with OCA and RP (Fig. 2 J and K). All findings were similar between the right and the left eye.Fig. 2. Imaging Comparison with a Control with Retinitis Pigmentosa and a Control with Albinism.On the left column; retinal imaging of a 49-year-old male with TYR-albinism and PDE6A-retinitis pigmentosa (RP). Middle column depicts retinal imaging of a 47-year-old female with the same homozygous pathogenic PDE6A-RP variant and isolated RP. Right column depicts retinal imaging of a 49-year-old male with TYR-albinism. (A–C) Widefield pseudocolor fundus images. (D–E) fundus autofluorescence (FAF) images with (G–I) FAF images of the macula at greater magnification, and (J–L) corresponding transfoveal optical coherence tomography (OCT). (G) and (H) show similar pattern of signal changes at the arcades in both patients and vessel attenuation, with the patient with albinism having also visible choroidal vasculature in the periphery. In contrast, patients with isolated albinism do not have the areas of decreased signal on FAF (I), but have foveal hypoplasia on OCT (L). (J) OCT showed foveal hypoplasia and disrupted ellipsoid zone for the patient with albinism and RP. The patient with isolated RP had better preserved foveal ellipsoid zone compared to the patient with albinism and RP, and ERM. RP: retinitis pigmentosa.Fig. 2
We also identified a matching 48 year old male control in the MEH database, compound heterozygous for the same TYR variant c.1467dup, p. (Ala490Cysfs*20), and the c.1366+4A > G TYR variant. Age of disease onset was 5 years old, presenting with nystagmus. BCVA was 0.3 LogMAR (20/40) and 0.5 LogMAR (20/60). Patient had 360° iris transilluminations. DFE showed foveal hypoplasia, blonde fundi and choroidal show, to a lesser extent than the patient with concurrent RP (Fig. 2C). Widefield FAF showed mostly preserved signal (Fig. 2F), and the macula showed loss of normally decreased signal (Fig. 2 I), a finding also present in our case with OCA and RP (Fig. 2 G). OCT showed better preserved foveal EZ compared to the patient with OCA and RP, and the patient with isolated RP (Fig. 2 L). All findings were similar among the right and the left eye.
Discussion
6
OCA is characterized by the reduction or absence of melanin in the skin, hair, and eyes, and consequent photosensitivity, high risk of skin cancer, and reduced visual acuity and nystagmus. The non-syndromic OCA is inherited in an AR manner and is mainly due to disease-causing variants in four genes: TYR (OCA1), OCA2 (OCA2), TYRP1 (OCA3), and SLC45A2 (OCA4).4 The prevalence of OCA1 is 1:40,000 (ORPHA79431). The TYR (MIM 606,933) gene encodes tyrosinase, which participates in the catalysis of the conversion of tyrosine to melanin. OCA caused by variants in the TYR gene is divided clinically into two types: type IA is characterized by a complete lack of tyrosinase activity due to the production of an inactive enzyme, and type IB is characterized by reduced activity of tyrosinase. Our patient was homozygous for the TYR gene c.1467dup, p. (Ala490Cysfs20) variant that generates a frameshift resulting in a premature stop codon in the last exon. This is not expected to result in nonsense mediated decay, although it is predicted to truncate the protein, and may result in disrupted protein function. In the literature, this variant has been reported as heterozygous together with other disease causing TYR variants, causing OCA.5, 6, 7, 8 It is submitted to ClinVar (variation ID 3803) and is listed in the Albinism Database. The patient met almost all the previously suggested clinical criteria for the diagnosis of albinism including; grade 4 foveal hypoplasia, ocular hypopigmentation (both fundus and iris), nystagmus, and hypopigmentation of skin and hair.2 No ERG was undertaken and no optic nerve misrouting can be proven, which is a limitation of our study.
The PDE6A gene (OMIM *180,071) encodes cGMP-specific phosphodiesterase 6A of the retinal rod cells. This protein is heterotetrametric and consists of alpha, beta (PDE6B, OMIM *180,072), and 2 gamma subunits (PDE6G, OMIM *180,073), and is involved in the processing, transmission and amplification of the visual signal and causes autosomal recessive RP.1 PDE6A c.304C > A, p. (Arg102Ser) has previously been reported both in compound heterozygous and homozygous.9, 10, 11, 12 The codon appears to be highly conserved. We previously reported a missense variant affecting the same codon in the PDE6C gene causing achromatopsia.13 *PDE6B-*RP is the target of gene therapy trial (NCT03328130),14 with the results of the trial being relevant to future therapeutic prospects for PDE6A-RP.
Our patient did not exhibit the classic phenotypic features of RP,15 due to the concurrent presentation of OCA. The worsening of his vision was not expected in the clinical course of OCA. High degree of clinical suspicion is needed for patients with rare diseases for possible concurrent pathology. The FAF and OCT imaging were the most informative for the concurrent PDE6A-RP, even though the changes were subtle and the imaging quality was poor due to nystagmus. The vision of our patient was worse than age matched patients with either isolated *PDE6A-*RP or isolated TYR-OCA, likely due to the dual pathology. We have previously reported similar findings of worse outcomes with dual pathology for patients with albinism and proliferative diabetic retinopathy.16 Similarities in the FAF phenotypes can be tracked among the three phenotypes of the current report (Fig. 2). The patient with OCA and RP had areas of decreased signal similar to the patient with RP along the arcades, and loss of the normally decreased foveal signal similar to the patient with isolated OCA. Our patient is unlikely to benefit from any gene specific treatment in the future due to his dual pathology. Vision rehabilitation and cataract surgery may help maximize his potential acuity.17
Conclusions
7
We present a well-documented molecularly confirmed case of a patient with TYR-OCA and PDE6A-RP. The case highlights the need of clinical awareness for rare diseases and the usefulness of genetic testing.
Patient consent
8
Written consent to publish those cases has not been obtained. This report does not contain any personal identifying information.
Funding
None.
Authorship
9
All authors attest that they meet the current ICMJE criteria for authorship.
CRediT authorship contribution statement
Michalis Georgiou: Methodology, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. Shaima Awadh Hashem: Formal analysis, Data curation. Michel Michaelides: Writing – review & editing, Investigation, Data curation. Joseph G. Chacko: Writing – review & editing, Writing – original draft, Supervision, Data curation, Conceptualization. Sami H. Uwaydat: Writing – review & editing, Writing – original draft, Supervision, Investigation.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Georgiou M.Robson A.G.Fujinami K.Phenotyping and genotyping inherited retinal diseases: molecular genetics, clinical and imaging features, and therapeutics of macular dystrophies, cone and cone-rod dystrophies, rod-cone dystrophies, leber congenital amaurosis, and cone dysfunction syndromes Prog Retin Eye Res 202410124410.1016/j.preteyeres.2024.10124438278208 · doi ↗ · pubmed ↗
- 2Kruijt C.C.de Wit G.C.Bergen A.A.Florijn R.J.Schalij-Delfos N.E.van Genderen M.M.The phenotypic spectrum of albinism Ophthalmology 1252018195319603009835410.1016/j.ophtha.2018.08.003 · doi ↗ · pubmed ↗
- 3Abou Tayoun A.N.Pesaran T.Di Stefano M.T.Recommendations for interpreting the loss of function PVS 1 ACMG/AMP variant criterion Hum Mutat 392018151715243019204210.1002/humu.23626 PMC 6185798 · doi ↗ · pubmed ↗
- 4Grønskov K.Ek J.Brondum-Nielsen K.Oculocutaneous albinism Orphanet J Rare Dis 22007431798002010.1186/1750-1172-2-43PMC 2211462 · doi ↗ · pubmed ↗
- 5Norman C.S.O'Gorman L.Gibson J.Identification of a functionally significant tri-allelic genotype in the Tyrosinase gene (TYR) causing hypomorphic oculocutaneous albinism (OCA 1B)Sci Rep 7201744152866729210.1038/s 41598-017-04401-5PMC 5493628 · doi ↗ · pubmed ↗
- 6King R.A.Pietsch J.Fryer J.P.Tyrosinase gene mutations in oculocutaneous albinism 1 (OCA 1): definition of the phenotype Hum Genet 11320035025131368036510.1007/s 00439-003-0998-1 · doi ↗ · pubmed ↗
- 7Hutton S.M.Spritz R.A.A comprehensive genetic study of autosomal recessive ocular albinism in Caucasian patients Invest Ophthalmol Vis Sci 4920088688721832670410.1167/iovs.07-0791 · doi ↗ · pubmed ↗
- 8Grønskov K.Ek J.Sand A.Birth prevalence and mutation spectrum in Danish patients with autosomal recessive albinism Invest Ophthalmol Vis Sci 502009105810641906027710.1167/iovs.08-2639 · doi ↗ · pubmed ↗