A Tumor-Specific Molecular Network Promotes Tumor Growth in Drosophila by Enforcing a Jun N-Terminal Kinase–Yorkie Feedforward Loop
Indrayani Waghmare, Karishma Gangwani, Arushi Rai, Amit Singh, Madhuri Kango-Singh

TL;DR
This study identifies a molecular network in fruit fly tumor cells that promotes tumor growth through a feedback loop involving JNK and Yki, which could help in understanding and treating human cancers.
Contribution
The paper discovers a tumor-specific network involving Wg, Dronc, JNK, and Yki that drives tumor growth in Drosophila.
Findings
RasV12,scrib− tumor cells show increased Wg, Dronc, JNK, and Yki signaling required for tumor growth.
A Wg–Dronc–Yki–JNK feedback loop is specifically activated in polarity-impaired tumor cells.
The identified network is evolutionarily conserved and may inform cancer treatment strategies.
Abstract
Cancer genomics and transcriptomics have revealed genes and pathways altered in several cancers. These studies have provided valuable information about cancer cells, their origin, oncogenic processes, and signaling pathways. An emerging challenge is to further characterize activity levels and interactions of pathways to develop a deeper understanding of how specific alterations in these interactions promote tumor growth. Drosophila with its sophisticated genetics has proved valuable in studying cooperative oncogenesis. Using Drosophila tumor models, we report a tumor cell-specific network comprising four pathways. We show that Wingless and effector caspase Dronc direct a signal amplification loop involving JNK and Hippo-effector Yorkie (Yki). Our studies provide evidence from in vivo studies regarding the organization of tumor-promoting oncogenic pathways, which may be useful in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
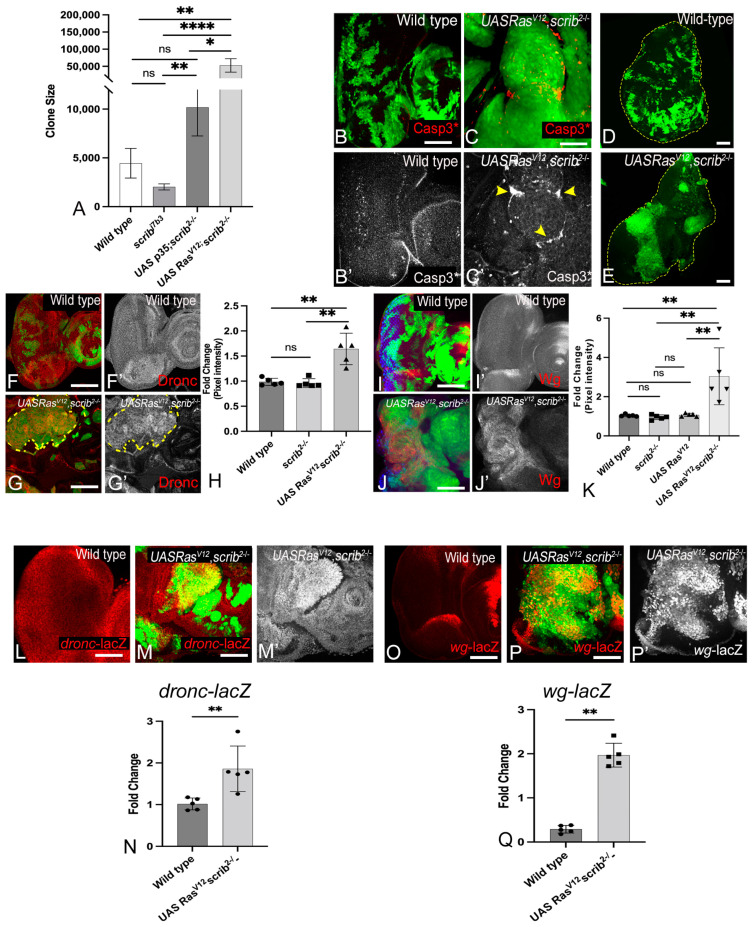 Figure 1
Figure 1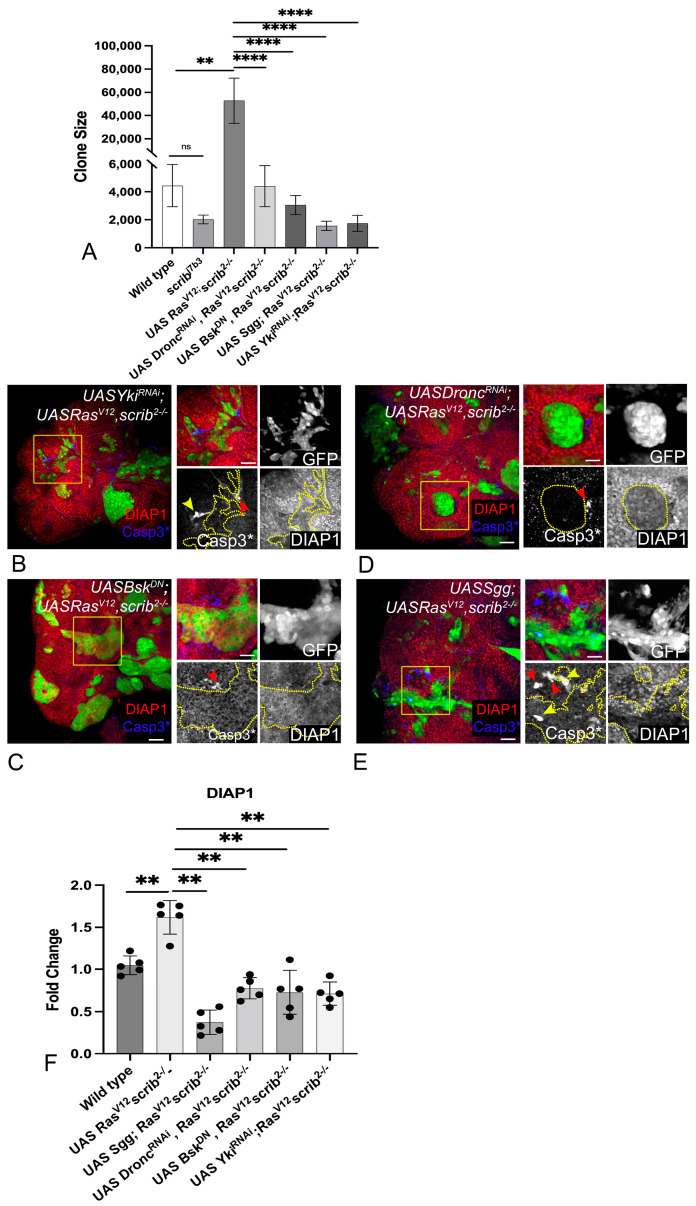 Figure 2
Figure 2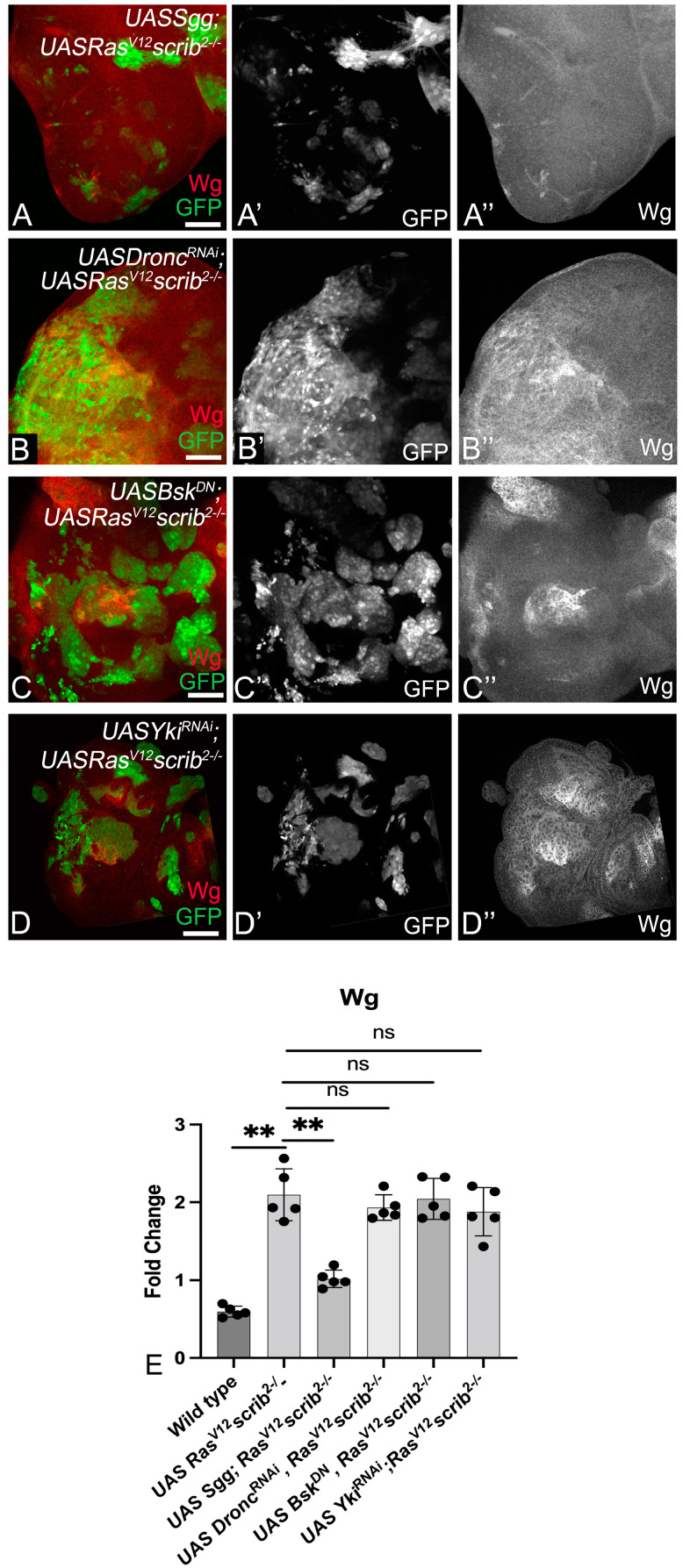 Figure 3
Figure 3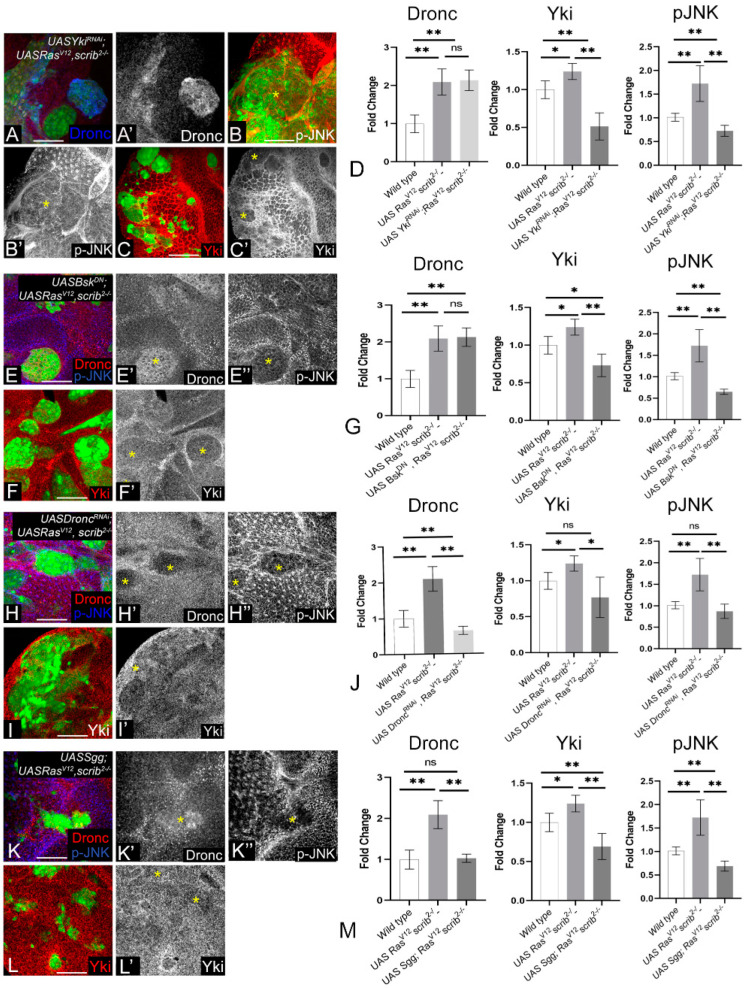 Figure 4
Figure 4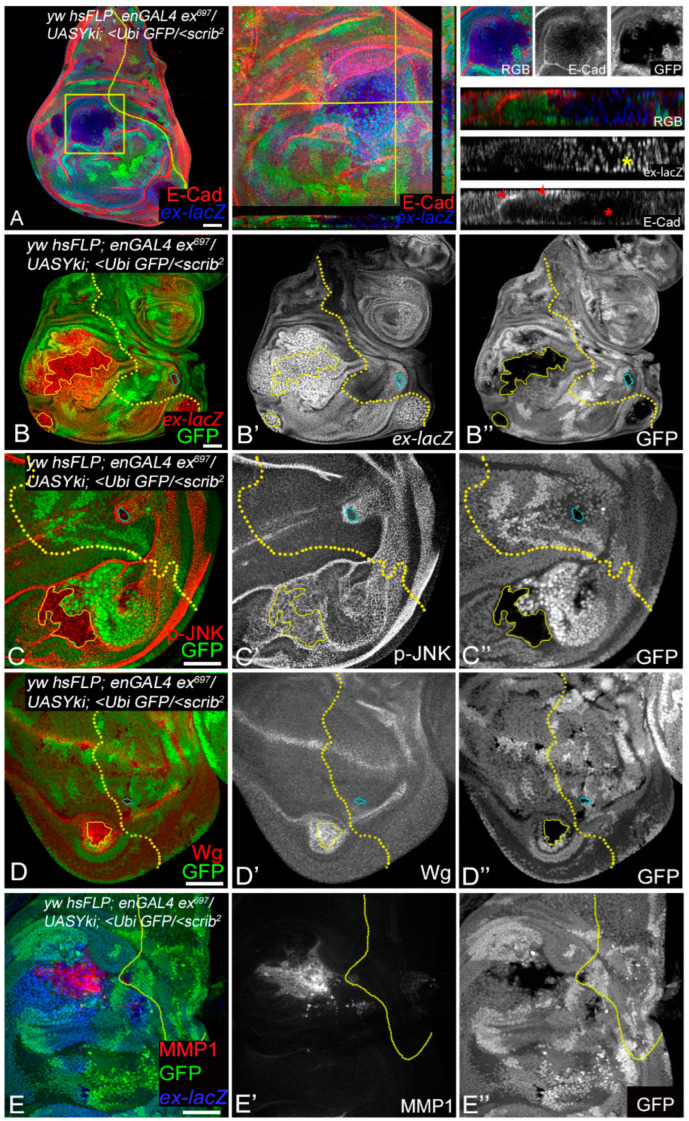 Figure 5
Figure 5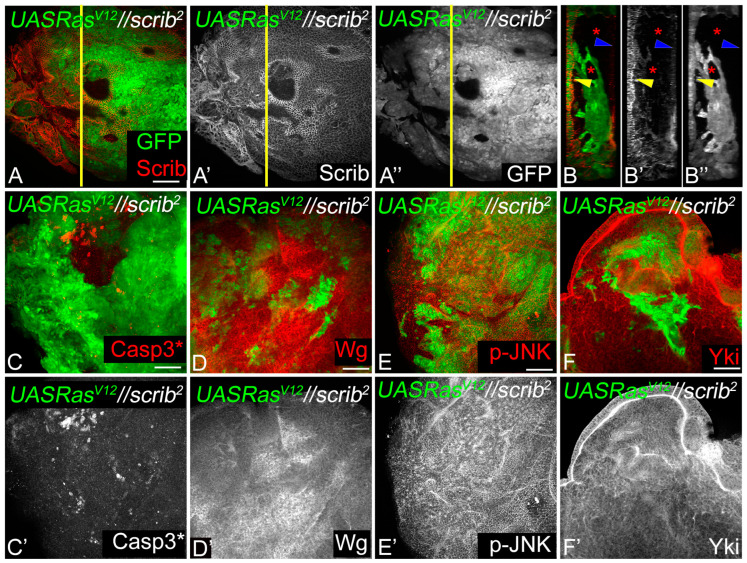 Figure 6
Figure 6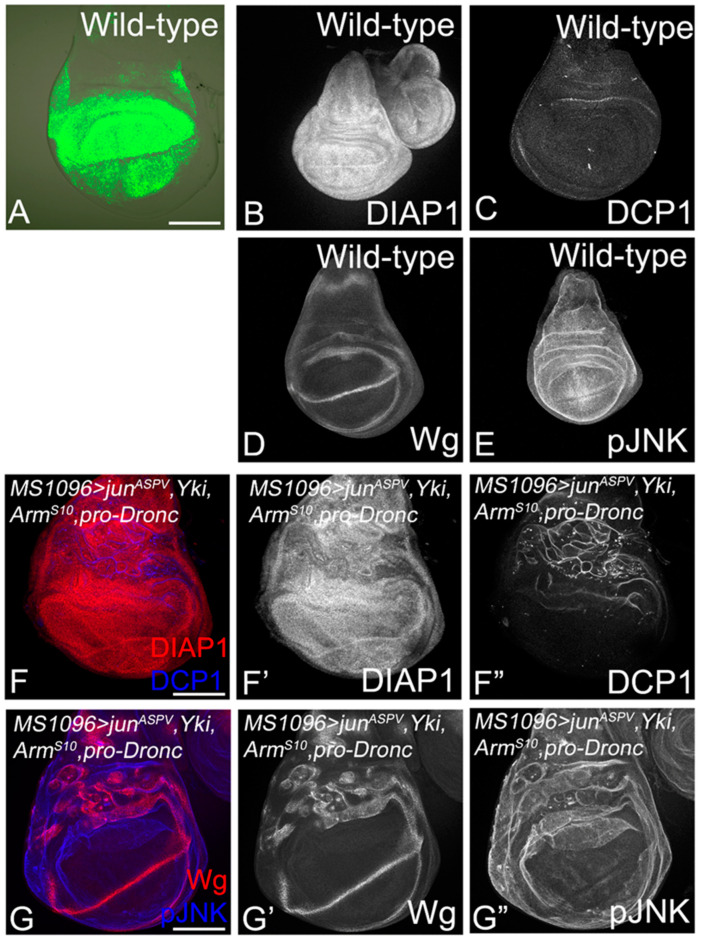 Figure 7
Figure 7- —University of Dayton
- —National Institutes of Health
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHippo pathway signaling and YAP/TAZ · Microtubule and mitosis dynamics · Cellular Mechanics and Interactions
1. Introduction
Over the past decade, genomic, transcriptomic, and proteomic data from several cancers have revealed the genetic alterations and changes in gene expression and epigenetic modifications linked to several cancers [1,2,3,4]. These approaches have revealed the extensive differences between normal and cancer cells, as well as the effects of cancer on multiple genes and signaling pathways [3]. Although these studies have provided valuable information about cancer cells, their origin, oncogenic processes, and signaling pathways, efforts now need to be directed at further characterizing tumors. For example, which proteins are expressed in the same cancer cell and are likely to physically interact? Which signaling pathways play decisive roles in promoting cell proliferation, survival, and metastasis in the different stages of cancer? Does the intricate distribution or clustering of ligand receptors affect cell–cell interactions [5]? For this, complementary approaches with in vivo models that allow manipulation of multiple genes (that represent the key cancer driver mutations) are required.
Drosophila, the common fruit fly, represents an efficient model organism to modulate the expression of multiple genes to study the signaling crosstalk that promotes tumor growth and progression [6,7]. Models of cooperative oncogenesis in Drosophila, such as clonal tumors induced by the activation of oncogenic Ras (Ras^V12^) in polarity-deficient scribble (scrib) [Ras^V12^ scrib^−^], lethal giant larva (lgl), or discs large (dlg) mutant cells, show classic hallmarks of aggressive cancer growth exemplified by increased proliferation rate, reduced apoptosis and differentiation, and metastasis [8,9,10,11]. These models have been instrumental in establishing the links between the deregulation of cell polarity and increased signaling from Jun N-terminal kinase (JNK) and Yorkie (Yki), effectors of two key signaling pathways that regulate cell proliferation and apoptosis [12,13,14,15,16]. However, the upstream mechanisms by which JNK and Yki activation promote aggressive metastatic growth during oncogenic cooperation remain poorly defined.
We investigated the changes in signaling interactions to decipher how oncogenic cooperation [Ras^V12^,scrib^−^] promotes tumor growth. Herein, we show that the Ras^V12^,scrib^−^ tumors grow aggressively by upregulating a previously unidentified molecular network comprising the Wingless (Wg, the Drosophila homolog of mammalian Wnt1), the effector caspase Dronc (Drosophila homolog of mammalian Caspase 9), JNK, and Yki (Drosophila homolog of Hippo effectors YAP/TAZ in mammals). The upregulation of these signals promotes the growth of Ras^V12^,scrib^−^ tumors, and the depletion of these signals reverses these phenotypes. These signals are well-known for their roles in patterning and growth control [17,18,19,20,21] and intercellular interactions like cell competition [22,23,24,25,26,27,28]. We show that during oncogenic cooperation, these signals act together in a tumor cell-specific signaling network, in which Wg acts upstream of Dronc and regulates JNK and Yki. JNK activity changes from pro-apoptosis to pro-proliferation due to a paradoxical signaling switch that ultimately promotes Yki activity. We demonstrate that in Ras^V12^,scrib^−^ tumor cells, Yki, in turn, upregulates JNK activity, causing the robust activation of both JNK and Yki. This bidirectional regulatory interaction results in the formation of a self-reinforcing JNK-Yki positive feedback signal-amplification loop downstream of Wg and Dronc. We show that this molecular network is sufficient to induce tumorigenesis in other contexts of oncogenic cooperation (such as en > Yki, scrib^−^, or Ras^V12^//scrib^−^ [interclonal tumor model [10]]). Taken together, our data strongly support the functional significance of molecular networks rather than individual signals in cancer cells and provide novel insights into the molecular pathways and paradoxical signals that play a key role in tumor growth and progression.
2. Materials and Methods
2.1. Fly Stocks
The studies described here are conducted on Drosophila melanogaster, the common fruit fly. All flies were reared in a standard cornmeal–agar–molasses medium containing Tegosept and propionic acid. All fly stocks used in this study are described in Flybase (http://flybase.bio.indiana.edu) accessed on 1 March 2024.
The following stocks were used in this study: Canton-S, FRT82B (BL#5619), FRT82B scrib^j7b3^ (DGRC#111422), FRT82B scrib^2^ [29], FRT82B scrib^2^ TubGAL-80 (this study), diap1-lacZ (BL#12093), dronc^1.7kb^-lacZ [30], ex^679^-lacZ [31], wg-lacZ [32], AyGAL4, UAS-GFP [33], en-GAL4 (BL#1973), UAS-GFP (BL#1521), FRT82B Ubi-GFP (BL#5188), FRT82B Tub-GAL80 (BL#5135), MS1096-Gal4 (BL#8860), UAS-P35 (BL#5072), UAS-Ras^V12^ (BL#4847), UAS-sgg^S9A^ (BL#5255), UAS-Bsk^DN^ (BL#6409), UAS-dronc^RNAi^ (gift from A. Bergmann, 8091R-1 from NIG-Fly (http://www.shigen.nig.ac.jp/fly/nigfly/index.jsp) accessed on 1 March 2024), UAS-Yki^N+CRNAi^ [34], UAS-proDronc (gift from A. Bergmann), UAS-jun^aspv^ [35], UAS-Arm^S10^ (BL#4782), and UAS-Yki [36].
2.2. Generation of Somatic Clones
To make MARCM clones, we crossed eyFLP or UbxFLP; AyGAL4 UASGFP; FRT82B Tub-GAL80 virgins with males of the appropriate genotypes. Heat shock-mediated “Flp-out” clones were generated by giving a 7 min heat shock at 37 °C to second instar larvae generated by crossing AyGAL4 UASGFP flies with UAS Yki. “Flp-out” clones were also generated by crossing yw hsFLP; enGAL4 ex^697^-lacZ; FRT82B UbiGFP flies with UAS Yki; FRT82B scrib^2^ flies (for en > Yki; scrib^−^ clones Figure 5). Interclonal oncogenic cooperation studies were performed by crossing yw eyFLP; AyGAL4 UASGFP; FRT82B Tub-GAL80 scrib^2^ virgins with yw hsFLP; +; UASRas^V12^ FRT82B males. All experiments were performed at 25 °C unless otherwise specified.
2.3. Immunohistochemistry and Image Acquisition
Immunohistochemistry was performed following our published protocol [37]. Briefly, imaginal discs from wandering 3rd instar larvae were dissected in 1XPBS (phosphate buffered saline), fixed in 4% paraformaldehyde (PFA) for 20 min (min) at room temperature (RT), washed 3 × 10 min each in 1XPBST (1XPBS + 0.2% Triton X-100), and blocked in PBSTN (PBST+ 2% Normal donkey serum) for 1 h before incubation with primary antibody at 4 °C overnight. The samples were then washed 3 × 10 min each in 1XPBST, incubated in an appropriate secondary antibody for two hours at RT, washed in 1XPBST for 20 min, and mounted in Vectashield (Vector Laboratories Inc., Burlingame, CA, USA).
The following primary antibodies were used: rabbit anti-cleaved Caspase 3 (1:250), rabbit anti-DCP1 (1:100), and rabbit anti-pJNK (1:250) from Cell-Signaling Technology, Danvers, MA, USA, mouse anti-Wingless (1:100), mouse or rabbit anti-beta-galactosidase (1:100), mouse anti-MMP1 (1:250), and rat anti-E-Cadherin (dCAD2, 1:100) from DSHB, Iowa, IA, USA, guinea pig anti-Dronc (1:400, from Dr. H. D. Ryoo), mouse anti-DIAP1 (1:250, from Dr. B. Hayes), and rabbit anti-Yorkie (1:400, from Dr. K. Irvine). The secondary antibodies were donkey Fab fragments conjugated to Cy3 or Cy5 against rabbit, mouse, rat, or guinea pig hosts (Jackson ImmunoResearch Labs, West Grove, PA, USA).
Image acquisition: We used an Olympus Fluoview 1000 confocal microscope to acquire projections of Z-stacks of confocal images. To enable comparison between samples, all images in an experimental group were acquired at identical magnification and similar imaging conditions. The final figures were made using Adobe Photoshop CS6.
2.4. Statistical Analyses
Statistical analyses were performed using Microsoft Excel 2013. The magnetic lasso tool was used for clone size comparison (Figures 1A and 2A Wild type n = 11, rest n = 25). Mean pixel values of clone area were obtained using the Histogram function in Photoshop CS6 and analyzed using the non-parametric Mann–Whitney test (two-tailed), assuming statistical significance at p < 0.05. Signal intensity levels for both inside (GFP-positive) and outside (GFP-negative), the clones were obtained using the histogram function in Adobe Photoshop CS6 (n = 5). The average wild-type values were used to find the normalization factor to determine and calculate differences in intensity levels between wild-type, scrib^−^, Ras^V12^, and Ras^V12^; scrib^−^ clones for all antibodies tested. Bar scatterplots were then generated to show fold-change in Dronc, Wg, dronc^1.7kb^-lacZ, wg-lacZ, diap1-lacZ, pJNK levels (Figure 1 and Figure S1), and DIAP1 (Figure 2F and Figure S3). Similarly, the normalization factor was calculated using wild-type values (GFP-negative areas) to assess changes in the expression of Wg (Figure 3), Yki, pJNK, and Dronc (Figure 4). Statistical significance (p < 0.05) was tested using a non-parametric Mann–Whitney test (two-tailed), and all graphs were plotted using GraphPad Prism9.0.
2.5. qRT-PCR
RNA was extracted from eye imaginal discs from wandering third instar larvae. At least 40–50 discs were collected per genotype in TRIzol. The ZYMO RNA Clean and Concentrator Kit was used for RNA extraction. To prepare cDNA, 200 ng/μL RNA/sample was reverse transcribed using the cDNA synthesis kit (Cytiva, Danaher Corporation, Washington, DC, USA). qRT-PCRs were performed in triplicate per sample using the iQ SYBER Green Supermix (Bio-Rad Laboratories, Hercules, CA USA) on an iCycler iQ™ Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA USA). Three biological and three technical repeats were performed for each experiment, and data were analyzed using the ddCt method.
The following primers (IDT) were used:
wg: Forward primer*: 5′* CGT CAG GGA CGC AAG CAT A-3′
Reverse Primer*: 5′* ATT GTG CGG GTT CAG TTG G-3′
dronc: Forward primer*: 5′* CGA TGG ATC TGT GGT CGA TAT G-3′
Reverse Primer*: 5′* GGC TTC GCT CGT CTT CTT TA-3′
Gapdh: Forward primer*: 5′* TAA ATT CGA CTC GAC TCA CGG T-3′
Reverse Primer*: 5′* CTC CAC CAC ATA CTC GGC TC-3′
Statistical analyses were performed using the non-parametric Mann–Whitney test (two-tailed), assuming statistical significance at p ≤ 0.05. Graphs were plotted using GraphPad Prism9.0.
3. Results
3.1. RasV12,scrib− Cells Grow Robustly and Induce JNK, Yki, Dronc, and Wg
To investigate the changes in intercellular interactions that promote tumor growth, we made ey-FLP- and Ubx-FLP-induced MARCM clones [GFP positive] in the eye (Figure 1A–C) and wing discs (Figure 1D,E) respectively and monitored clone size to assess tumor growth (Figure 1A). Compared to wild-type (Figure 1A), scrib^−^ clones grew poorly (Figure S1A) [38]. Given that scrib^−^ clones are competed out due to cell competition-mediated apoptosis [12,38], we blocked apoptosis by overexpressing UASp35 (Figure S1B), a pan-Caspase inhibitor [39]. The inhibition of apoptosis improved the growth of scrib- clones (scrib^−^, p35, Figure 1A and Figure S1C); however, the discs remain monolayered, and clones did not form invasive tumors (Figure S1C,C’ and YZ sections in Figure S1C”). In contrast, the coexpression of oncogenic Ras (UASRas^V12^) in scrib^−^ cells (Ras^V12^,scrib^−^) resulted in robust aggressive and invasive tumors [8] that grew severalfold compared to wild-type, scrib^−^, or scrib^−^, p35 clones (Figure 1A). These data suggest that a paradoxical switch changes the ability of scrib^−^ cells, and inter-cellular interactions may play a critical role in these observed effects. Next, to understand the nature of the intercellular interactions that may underlie the differences in clone sizes, we tested cell death by using an antibody against activated Caspase 3 [Casp3*]. In comparison to wild-type clones (Figure 1B,B’), Ras^V12^,scrib^−^ clones show cell death both inside and outside the clone, and interestingly, the bulk of the observed cell death was induced in wild-type cells surrounding the clone (Figure 1C,C’ and Figure S1B–B”). Similarly in the wing discs, compared to wild-type (Figure 1D), Ras^V12^,scrib^−^ clones grew into large invasive tumors (Figure 1E and Figure S1D). Taken together, these data suggested that the Ras^V12^,scrib^−^ clones grow robustly. Therefore, as a next step, we tested if JNK, Yki, Dronc, and Wg, markers that have previously been linked to cell survival and proliferation during cell competition [22,23,24,40] were also affected in the Ras^V12^ scrib^−^ tumors.
JNK and Yki are known to interact in a context-dependent manner [12,13,14,41], and increased JNK or Yki activity has been linked to tumor growth [12,42,43,44,45]. In contrast, the elimination of scrib^−^ cells by cell competition involved JNK-dependent suppression of Yki activity [12,38,46,47,48]. To test Yki activity, we employed the Yki-reporter, diap1-lacZ, which is expressed ubiquitously in wild-type (Figure S1E) discs. Increased Yki activity due to Yki overexpression in “Flp-out” (AyGAL4 > Yki) clones showed the robust induction of diap1-lacZ (Figure S1F,F’). In the Ras^V12^,scrib^−^ clones, diap1-lacZ was strongly induced (Figure S1G,G’). Interestingly, diap1-lacZ was also induced in peri-tumoral cells (non-cell autonomously in wild-type cells) abutting the Ras^V12^,scrib^−^ tumors (Figure S1G’). The normalized pixel intensity from the controls and experimental groups was quantified and showed a significant increase in Ras^V12^,scrib^−^ clones (Figure S1H). Compared to wild-type (Figure S1I,I’), pJNK was robustly induced in the Ras^V12^,scrib^−^ clones (Figure S1J,J’, quantified in Figure S1K). Thus, consistent with previous data, JNK and Yki activities were simultaneously upregulated in the Ras^V12^,scrib^−^ tumors. We then investigated the signals that converge onto Yki and JNK to promote the growth of Ras^V12^,scrib^−^ tumors.
Previous studies have shown that Dronc (the initiator caspase that induces apoptosis through activation of caspase 3) and Wg (the secreted ligand of the Wingless/Wnt pathway that acts as a mitogen) play critical roles in intercellular interactions like cell competition and compensatory proliferation [27,49,50,51]. Yki also transcriptionally regulates dronc and wg [52,53]. We tested levels of active Dronc using an antibody against Dronc^CA^, the activated form of Dronc [51], and Wg in the Ras^V12^,scrib^−^ clones (Figure 1). Dronc is expressed ubiquitously in wild-type imaginal discs (Figure 1F,F’, quantified in Figure 1H), and its levels remained unaffected in scrib^−^ cells in the wild-type background (Figure 1H). Therefore, wild-type Dronc levels may be sufficient for the elimination of scrib^−^ cells in the presence of elevated pJNK levels. In comparison, Dronc was strongly upregulated in Ras^V12^,scrib^−^ cells (Figure 1G,G’). Quantification showed that Dronc levels were significantly upregulated (1.7-fold) in Ras^V12^,scrib^−^ cells compared to either wild-type or scrib^−^ cells (Figure 1H). Taken together, these data suggested that Dronc plays a non-apoptotic role and promotes Ras^V12^,scrib^−^ tumor growth. In wild-type eye discs, Wg is expressed at the lateral margins anterior to the morphogenetic furrow (Figure 1I,I’, quantified in Figure 1K) [32]. This pattern of Wg expression was unaltered in scrib^−^ cells generated in the wild-type background (Figure 1K). In Ras^V12^,scrib^−^ clones, Wg was robustly induced (Figure 1J,J’). When compared to wild-type or scrib^−^ cells, Wg expression was significantly upregulated (2.7-fold) in Ras^V12^,scrib^−^ cells (Figure 1K), suggesting that the steep upregulation of Wg levels in Ras^V12^,scrib^−^ may itself promote super-competition by promoting proliferation through its mitogenic functions.
Next, we checked if dronc and wg transcription was affected in Ras^V12^,scrib^−^ cells using the dronc^1.7kb^-lacZ (Figure 1L–M’) and wg-lacZ (Figure 1O–P’) reporters, quantified in Figure 1N,Q, respectively. We found that compared to the wild-type (Figure 1L), dronc^1.7kb^-lacZ expression was robustly induced in the Ras^V12^,scrib^−^ clones (Figure 1M,M’). Similarly, the normal pattern of wg-lacZ expression in eye discs (Figure 1O) was disrupted due to ectopic induction of wg-lacZ in Ras^V12^,scrib^−^ clones (Figure 1O’). We confirmed the increased expression of dronc and wg in Ras^V12^,scrib^−^ clones using qRT-PCR (Figure S2). The upregulation of dronc and wg in the tumor cells suggests that these genes promote tumor growth, possibly by co-opting compensatory mechanisms in which dronc plays a non-apoptotic role and wg plays a mitogenic role. Taken together, these data suggest that four signals associated with intercellular interactions (Yki, JNK, Wg, caspases) were upregulated in aggressively growing Ras^V12^,scrib^−^ tumors.
3.2. RasV12,scrib− Tumors Require JNK, Yki, Dronc, and Wg for Growth
To understand if some or all of these signals played a key role in the aggressive growth of Ras^V12^,scrib^−^ tumors, we tested their requirement by independently downregulating these signals in Ras^V12^,scrib^−^ clones (Figure 2). To achieve this, we used well-established transgenes, for example, RNAi, to knockdown Dronc, which impairs caspase-mediated signaling [UAS-Dronc^RNAi^] [52], the dominant-negative form of Bsk, which is a potent suppressor of JNK signaling [UAS-Bsk^DN^] [38], UAS-Sgg^S9A^, which dominantly blocks Wg signaling [54], and UAS-Yki^N+CRNAi^, which causes the inactivation of Yki-mediated signaling [34].
A comparison of clone sizes revealed that individual downregulation of the four signaling pathways significantly decreased the growth of the Ras^V12^,scrib^−^ cells (Figure 2A–E). These findings suggested that the four signals are independently required for the aggressive growth of Ras^V12^,scrib^−^ tumors. Therefore, we next tested if loss of cellular fitness or interactions amongst these signals was an underlying cause for the change in tumor size.
3.3. Downregulation of JNK, Yki, Dronc, and Wg Impairs Cellular Fitness
We hypothesized that the reduction in clone size was due to altered fitness within Ras^V12^,scrib^−^ cells. First, we tested changes in DIAP1 levels (Figure 2B–E red, grey) in conditions where at least one of these four signals was downregulated (Figure 2B–F). As DIAP1 inhibits caspase activation, it protects cells from caspase-mediated apoptosis [55]. DIAP1 was ubiquitously expressed in wild-type cells (Figure S3A), downregulated in scrib^−^ cells (Figure S3B, yellow arrowheads), and robustly induced in Ras^V12^,scrib^−^ clones (Figure S3C). However, when Yki (Figure 2B), JNK (Figure 2C), Dronc (Figure 2D), or Wg (Figure 2E) were downregulated in Ras^V12^,scrib^−^ clones (Figure 2C–E, yellow outline in grey channels), DIAP1 was downregulated (quantified in Figure 2F). This finding suggests that Ras^V12^,scrib^−^ tumor cells had decreased survival and were susceptible to elimination by apoptosis (Figure 2B–E, blue, grey), when the tumor-promoting signals were downregulated. The downregulation of Yki (Figure 2B) or Wg (Figure 2E) resulted in apoptosis both inside (Figure 2B,E yellow arrowheads) and outside (Figure 2B,E, red arrowhead) the Ras^V12^,scrib^−^ clones, suggesting that Wg and Yki protect Ras^V12^,scrib^−^ tumors during competitive interactions. The downregulation of the JNK pathway (Figure 2C) or Dronc (Figure 2D) resulted in apoptosis at the clone boundary (Figure 2C,D, red arrowhead), but not within the clones, likely because these cell death regulators (JNK and Dronc) were suppressed in the clones. Taken together, these data suggested that the downregulation of the tumor-promoting signals impaired cellular fitness in Ras^V12^,scrib^−^ cells.
These observations were further supported in control experiments, where we made MARCM clones in which we depleted each component of the molecular network in otherwise wild-type cells (Figure S4). Wg is a target of its own signaling [56,57,58]. Therefore, we made UAS-Sgg^S9A^ control clones (Figure S3A,B) and confirmed that the endogenous expression of Wg was suppressed in UAS-Sgg^S9A^ clones using anti-Wg antibody (shown by the yellow arrow in Figure S4B”). The downregulation of Wg signaling (Figure S4B red, Figure S4B” grey) caused cell death (Figure S4A blue, Figure S4A’’’ grey) and the downregulation of DIAP1 (Figure S4A red, Figure S4A” grey). However, the levels of pJNK (Figure S4B blue, Figure S4B’’’ grey) remained unaltered. The depletion of Dronc blocked cell death (Figure S4C blue, Figure S4C’’’ grey) and did not significantly affect DIAP1 (Figure S4C red, Figure S4C” grey), Wg (Figure S4D red, Figure S4D” grey), or pJNK (Figure S4D blue, Figure S4D’’’ grey) expression. Blocking JNK signaling (Figure S4E,F) showed similar effects to those of the downregulation of Dronc on DIAP1 (Figure S4E red, Figure S4E” grey), cell death (Figure S4E blue, Figure S4E’’’ grey), Wg (Figure S4F red, Figure S4F” grey), or pJNK (Figure S4F blue, Figure S4F’’’ grey). The depletion of Yki (Figure S4G,H) resulted in the downregulation of DIAP1 (Figure S4G red, Figure S4G” grey) and a strong non-cell autonomous induction of cell death (Figure S4G blue, Figure S4G’’’ grey). No significant effects on Wg (Figure S4H red, Figure S4H” grey) or pJNK (Figure S4H blue, Figure S4H’” grey) were seen. Thus, Wg and Yki emerged as key signaling proteins among these four pathways.
3.4. Wg, Dronc, JNK, and Yki Form a Molecular Network in RasV12,scrib− Tumors
Next, we tested if Wg, Dronc, JNK, and Yki regulated one another to promote the aggressive growth of Ras^V12^,scrib^−^ tumors (Figure 3). The downregulation of Wg signaling in Ras^V12^,scrib^−^ cells (Figure 3A) resulted in small tumors and a reduction in Wg expression (Figure 3A red, A” grey). Interestingly, depleting Dronc (Figure 3B), JNK (Figure 3C), or Yki (Figure 3D) caused a significant decrease in clone size, but Wg was strongly induced in these clones (Figure 3B”–D”). It is notable that Wg is induced in both a cell-autonomous and non-cell-autonomous manner. Normalized signal intensity is quantified in Figure 3E. Thus, we identified that Wg acts upstream of Dronc, JNK, and Yki in the network of signals that promote Ras^V12^,scrib^−^ tumor growth.
To test if these signals interact with each other, we checked the effects of Yki depletion (Yki^RNAi^; Ras^V12^,scrib^−^, Figure 4A–D) on Dronc (Figure 4A) and pJNK expression (Figure 4B). The depletion of Yki (confirmed in Figure 4C red, C’ grey) did not affect the induction of Dronc (Figure 4A blue, A’ grey); however, pJNK expression was downregulated in these clones (Figure 4B red, B’ grey). We quantified the normalized mean grey values and plotted the fold change comparing wild-type to Ras^V12^,scrib^−^ and Yki^RNAi^; Ras^V12^,scrib^−^. Graphs show the significant downregulation of Yki and pJNK, whereas Dronc accumulation was not affected (Figure 4D). Next, we downregulated JNK signaling (Figure 4E–G) through the overexpression of a dominant negative form of basket (Bsk) in Ras^V12^,scrib^−^ (Bsk^DN^; Ras^V12^,scrib^−^), which results in the downregulation of pJNK (Figure 4E blue, E” grey), but Dronc expression remained upregulated in these clones (Figure 4E red, E’ grey). Interestingly, Yki expression was also depleted in Bsk^DN^; Ras^V12^,scrib^−^ cells (Figure 4F red, F’ grey). We plotted the normalized fold change in the expression of Dronc, Yki, and pJNK, which confirms these effects (Figure 4G). Interestingly, the downregulation of Dronc (Dronc^RNAi^; Ras^V12^,scrib^−^, Figure 4H–J) in Ras^V12^,scrib^−^ cells (Figure 4H red, H’ grey) showed the downregulation of JNK (Figure 4H blue, H” grey) and Yki (Figure 4I red, I’ grey). Quantification shows that compared to wild-type or Ras^V12^,scrib^−^, the significant downregulation of Yki and JNK is seen in Dronc^RNAi^; Ras^V12^,scrib^−^ clones (Figure 4J). The downregulation of Wg (Figure 4K–M) in Ras^V12^,scrib^−^ cells (Sgg^S9A^; Ras^V12^,scrib^−^) showed the downregulation of Dronc (Figure 4K red, K’ grey), pJNK (Figure 4K blue, K” grey), and Yki (Figure 4L red, L’ grey). Quantification of the effects of downregulation of Wg revealed significant downregulation of Dronc, Yki, and pJNK compared to Ras^V12^,scrib^−^ clones (Figure 4M). These data show that Dronc levels remain upregulated and comparable to Ras^V12^,scrib^−^ cells in combinations in which either Yki (Figure 4D) or JNK (Figure 4G) are downregulated. Thus, these data suggest that Dronc acts upstream of Yki and JNK in this signaling network. Furthermore, JNK and Yki regulate each other, suggesting that they may act in a feedforward loop downstream of Dronc. Overall, our data suggest that the growth of the Ras^V12^,scrib^−^ clones depended on a network involving Wg-dependent activation of Dronc, which controls a JNK-Yki mediated signal amplification loop that sustains high levels of JNK and Yki activities.
3.5. Cooperative Interactions Stimulate the Molecular Network and Tumor Growth
The preceding data led us to ask if oncogene activation, loss of polarity (scrib^−^), or cooperative interactions were important in stimulating the molecular network and tumor growth. The loss of polarity (scrib) alone is insufficient to induce aggressive growth in somatic clones (Figure 1 and Figure S1) [8,9,12,38]; therefore, we tested the importance of cooperative interactions on the induction of the Wg–Dronc–JNK–Yki network and tumor growth. We tested two scenarios in which the loss of polarity could synergize with oncogene activation.
In the first scenario, we generated scrib^−^ clones [GFP-negative clones in the FRT82B Ubi-GFP background generated by hs-FLP] in wing discs in which Yki was overexpressed [UAS-Yki] in the posterior compartment using en-GAL4 [en > Yki; scrib^−^] (Figure 5). In the eye or wing discs, scrib mutant clones are susceptible to elimination by cell competition due to the upregulation of JNK (Figure S5A–B’), downregulation of Yki activity, as seen through the suppression of diap1-lacZ (Figure S5C–D’ yellow arrowheads), and little to no effect on Dronc (Figure S5E) or Wg (Figure S5F) expression. Whereas, consistent with published data, the over-expression of Yki results in robust overgrowth due to the induction of Yki activity, leading to the upregulation of JNK [14,16], activation of DIAP1 (Figure S1F), ectopic induction of Wg [16], and suppression of Dronc [52]. Thus, the loss of scrib had clear and distinguishable effects from Yki overexpression (Figure 5 and Figure S5) on all four signals. These distinct effects allowed an ideal opportunity to test altered signaling caused during oncogenic cooperation by Yki overexpression in scrib mutant cells. We found that scrib^−^ clones grew to significantly larger sizes in the posterior (P) compartment in which Yki was overexpressed (Figure 5A–E) and showed reduced E-cadherin expression (Figure 5A, red, grey, red asterisk). Increased Yki activity in the posterior compartment was monitored by ex-lacZ expression (Figure 5B red, B’ grey), which also allows marking the anterior–posterior (A/P) boundary (shown in the yellow dotted line in Figure 5). In addition to the increased ex-lacZ staining observed in the posterior compartment due to yki overexpression, Yki activity in the scrib- cells in the posterior compartment was very strongly induced within and around the clones (Figure 5B, outlined by the yellow line). We called this dramatic increase in Yki activity “super-induction”. In these clones, pJNK (Figure 5C red, C’ grey), Wg (Figure 5D red, D’ grey), and MMP1 (Figure 5E red, E’ grey) were induced in and around the scrib^−^ clones, potentially leading to the establishment of the JNK–Yki loop and invasiveness by the induction of MMP1. The Yki, pJNK, and Wg signals spread to several cells outside the scrib^−^ clones, with Yki activity propagating the farthest (Figure 5B,B’). In contrast, the anterior (A) compartment scrib^−^ clones (Figure 5B–E blue outline) behaved like clones in the wild-type background (Figure S1A) and were eliminated through cell competition. A comparison of the P and A compartment-specific clones suggested that the JNK–Yki signal amplification loop was activated specifically in the tumor cells in the P compartment in which cooperative interactions occurred due to Yki expression in scrib^−^ cells.
In the second scenario, we tested if the Wg–Dronc–JNK–Yki network was activated during interclonal cooperation when Ras^V12^ and scrib^−^ cells were adjacent to each other (Ras^V12^//scrib^−^) [10]. Consistent with previous data, aggressive Ras^V12^ tumors (GFP-positive) were formed adjacent to smaller scrib^−^ clones (GFP negative, Figure 6A) [10]. Interestingly, the Ras*^V12^* clones extruded apically and showed a multi-layered invasive phenotype (Figure 6A–A”, Y-Z sections in Figure 6B–B”). The Ras^V12^ clones showed robust growth despite the induction of cell death at the clone boundary between Ras^V12^ (GFP-positive) and scrib^−^ (GFP-negative) (Figure 6C,C’) clones. The expression of Wg was robustly induced in the scrib^−^ clones (Figure 6D,D’), whereas pJNK levels appear upregulated in both scrib^−^ and Ras^V12^ clones (Figure 6E,E’) [10,38]. Yki expression showed higher nucleocytoplasmic distribution in the Ras^V12^ clones (Figure 6F,F’). Taken together, these data suggest that the robust growth of the Ras^V12^ clones was dependent on the interclonal signaling interactions with scrib^−^ clones and involved caspases, Yki, Wg, and JNK signaling. Overall, these two experimental approaches reaffirmed that the induction of the molecular network was intimately linked to tumor growth in diverse scenarios.
3.6. The Role of Apical–Basal Polarity in the Establishment of Tumor-Specific Molecular Networks
The Wg–Dronc–Yki–JNK network requirement in promoting tumor growth under various cooperative contexts raised the question if these four signals were sufficient to induce tumors in normal cells or if the loss of polarity is essential to form aggressive tumors. To test this, we first tested the effect of overexpressing Wg–Dronc–Yki–JNK in normal cells using the Gal4-UAS system in wing-imaginal discs. We used the wing hinge and pouch-specific MS1096-Gal4 (Figure 7A) to drive the expression of transgenes overexpressing Yki (UAS-Yki), Dronc (UAS-proDronc), JNK (UAS-jun^aspv^), and Wg (UAS-Arm^S10^) [MS1096 > Yki,pro-Dronc, jun^aspv^, Arm^S10^], which would result in the activation of all four signals (Figure 7). In wild-type wing discs, DIAP1 (Figure 7B) and pJNK (Figure 7E) are ubiquitously expressed, whereas Wg is expressed in the wing hinge, wing margin, and notum (Figure 7D). Wild-type discs show few randomly dying cells marked by DCP-1 (Figure 7C). Interestingly, the coexpression of these transgenes resulted in hyperplasia of the wing pouch and hinge region (Figure 7F,G). We observed moderate upregulation of DIAP1 in the wing pouch region (Figure 7F’) and increased cell death in the wing hinge region (Figure 7F”). Both Wg (Figure 7G,G’) and pJNK are induced in a patchy pattern in the dorsal hinge region (Figure 7G,G”). In control experiments, the overexpression of individual transgenes revealed that the overexpression of Yki (MS1096 > Yki) is capable of driving hyperplasia and the upregulation of DIAP1, Wg, and pJNK (Figure S6A–B”). The overexpression of pro-Dronc (MS1096 > proDronc) did not affect disc growth very strongly and showed moderate upregulation of DIAP1, cell death, Wg, and pJNK (Figure S6C–D”), suggesting that pro-Dronc plays a role in promoting survival, but not cell proliferation. Overexpression of jun^aspv^ (MS1096 > jun^aspv^) showed the most interesting effects by reducing the size of the wing pouch due to strong suppression of DIAP1 and the induction of cell death (Figure S6E–E”). Although Wg and pJNK are induced in this combination (Figure S6F–F”), the excessive cell death in the wing pouch does not support the growth of these discs. The activation of the Wg pathway through the overexpression of Arm^S10^ (MS1096 > Arm^S10^) resulted in the upregulation of DIAP1, Wg, and pJNK (Figure S6G–H”). In addition, mild to moderate levels of cell death were observed in the wing pouch (Figure S6G”). Taken together, these data suggest that although the coactivation of these transgenes results in increased growth or mild hyperplasia, the presence of normal wild-type cells with intact polarity does not allow for the establishment of the network that would drive the tumor growth.
4. Discussion
Oncogenic cooperation is a key mechanism for tumor development and progression. Cooperative interactions between oncogenic Ras and loss of scrib^−^ (Ras^V12^,scrib^−^) have been elegantly modeled using in vivo mosaic tumor models in Drosophila [8,9,12,38] and multiple mammalian cancer models [59,60,61,62,63]. We investigated how altered signaling and cell–cell interactions promote tumorigenesis during oncogenic cooperation. Previously ectopic upregulation of a network of transcription factors, e.g., Upd, JAK-STAT, AP-1, Myc, Ftz, Ets, Irbp18, Xrp1, slow border, and Vrille, were reported to promote the growth and invasiveness of the Ras^V12^,scrib^−^ tumors [64,65,66,67,68]. Data acquired from transcriptomics or RNA-seq have shown the presence of multiple transcription factors in the same genotype suggesting that several pathways and signals may be simultaneously active that culminate in the upregulation of several different transcription factors in tumor cells. These studies also confirm that several transcription factors are active based on the activation of their target genes. However, the potential molecular networks established remained unclear. Here we present evidence to show that a tumor cell-specific network is formed in Ras^V12^,scrib^−/−^ tumors in eye imaginal discs. This network is comprised of Yki, JNK, Wg, and caspases that act to promote fitness and aggressive growth (Figure 1 and Figure S1). Our findings from the eye discs are relevant to other epithelia, like the wing discs as the tumor-specific network, is recapitulated in the en > Yki; scrib^−^ wing tumors (Figure 5).
Of these signals, JNK is very well documented as a modulator of Yki activity in the context of cell competition, compensatory proliferation, regeneration, and neoplastic tumors [12,14]. JNK is a pivotal stress-responsive kinase that promotes malignant transformation and metastasis of tumors [38,45]. JNK has also emerged as a key paracrine signal that links apoptosis to carcinogenesis [12]. In scrib^−^ cells, JNK signaling suppresses Yki activity and promotes cell competition [12,69]. Concomitantly, JNK signaling causes the non-cell-autonomous propagation of Yki in the cells surrounding scrib^−^ clones and promotes compensatory proliferation [12,69]. Thus, in scrib^−^ clones in the wild-type background, JNK (Figure S5B) and Yki (Figure S5C,D) activities are induced in two distinct cell populations, and wild-type levels of Dronc (Figure S5E) or Wg (Figure S5F) are sufficient to support the cell competition-mediated elimination of scrib^−^ cells. Work from our lab and others showed that the suppression of cell death in scrib^−^ cells led to increased proliferation and the loss of differentiation, but not tumorigenesis (Figure S1A,C) [70,71]. In contrast, we found that these signaling interactions are significantly altered in Ras^V12^,scrib^−^ cells, as Wg, Dronc, JNK, and Yki are all robustly upregulated (Figure 1 and Figure S1), suggesting a non-apoptotic tumor growth-promoting role for JNK and Dronc and growth-promoting mitogenic roles for Yki and Wg. Using reporter assays for dronc and wg, we found that Ras^V12^,scrib^−^ cells show increased transcription of dronc (Figure 1L–N) and wg (Figure 1O–Q), which was confirmed using qRT-PCR (Figure S2). In addition, increased JNK activity using phospho-specific JNK antibody and Yki activity by diap1-lacZ reporter were confirmed in the Ras^V12^,scrib^−^ clones (Figure S1). These findings are consistent with reports that JNK signaling activity is converted from anti- to pro-tumor growth through the downregulation of Hippo signaling and ultimately leads to Yki activation [14,41]. Both Yki/YAP and JNK are linked to Wg signaling, as Wg is induced in a JNK-dependent manner during regenerative growth, tumorigenesis, and compensatory proliferation [51,72,73] and interacts with Yki and the Hippo pathway during organ development and tumorigenesis [74,75,76]. In addition, dysregulation of the Hippo, JNK, or Wg pathway is also linked to the activation of caspase-mediated apoptosis, and recently, mild caspase induction was shown to promote tumor growth [77,78,79]. Thus, the identification of this molecular network is significant in the context of inter-cellular interactions that promote tumor growth.
Previous studies have shown a role for the JNK and Hippo pathway in Ras^V12^ scrib^−^ clones [38,41]. In our study, an assessment of the roles of JNK, Yki, Dronc, and Wg in Ras^V12^,scrib^−^ tumorigenesis revealed several interesting insights. First, we found that JNK, Yki, Dronc, and Wg were all required for the aggressive growth of Ras^V12^,scrib^−^-induced tumors, as the downregulation of these signals individually resulted in a significant reduction in tumor growth (Figure 2A). Second, we observed that in the absence of tumor-promoting signals, Ras^V12^,scrib^−^-induced tumors show reduced survival and fitness (Figure 2). Third, we identified that the four tumor-promoting signals form a tumor cell-specific signaling module in which Wg acts upstream of Dronc, which, in turn, acts upstream of a JNK–Yki-mediated positive feedback signal amplification loop (Figure 3 and Figure 4). Signal amplification of Yki and JNK activities caused by the JNK–Yki positive feedback loop plays a key role in promoting tumorigenesis in Ras^V12^,scrib^−^ cells. Fourth, we confirmed that in other instances of oncogenic cooperation (Ras^V12^//scrib^−^), this signaling module can be recapitulated (Figure 5 and Figure 6). Fifth, the upregulation of JNK and Yki in normal epithelial cells with intact polarity is not sufficient to induce this network and tumor growth (Figure 7 and Figure S6), which is consistent with other instances of developmental interactions between Yki and JNK in polarized epithelia [80,81]. Taken together, our studies reveal that increased cellular fitness promoted by a molecular network comprising Wg, Dronc, JNK, and Yki may indeed be a mechanism co-opted for aggressive tumor growth. Furthermore, the overexpression of pathway components in the normal epithelia of wing discs revealed that the signal overactivation levels and the apical-basal polarity context are extremely important. This is because mild hyperplasia can occur by driving the individual transgenes, but robust overgrowth is not seen in cells with intact polarity. Further, for the JNK pathway, a threshold is critical, as stress-induced cell death occurs both when levels of JNK reach above or below the homeostatic levels (Figure 7 and Figure S6).
Another possible mechanism is the differential success of cancer and neighboring normal cells in competing for survival and other extracellular signals in terms of actively internalizing or limiting the extracellular spread of critical signals. In this context, we observed steep differences in the non-cell-autonomous spread of Yki activity among scrib^−^ (Figure 5 and Figure S5C), Ras^V12^ scrib^−^ (Figure S1G,G’), and en > Yki; scrib^−^ (Figure 5B) cells. Non-cell-autonomous Yki activity spreads to 3–5 cells in scrib^−^ cells, several cells (8–10) in en > Yki; scrib^−^, and only a few cells (~2) around the Ras^V12^,scrib^−^ clones. Further, cell-autonomous and non-cell-autonomous cell death is observed on the depletion of Yki (Figure 2B), suggesting that cells with impaired Yki expression are vulnerable to elimination by unidentified mechanisms. Interestingly, both cell-autonomous and non-cell-autonomous expression of Wg is seen when the components of the Wg–Dronc–JNK–Yki network are perturbed (Figure 3). Thus, limiting the extracellular spread of key signaling components like Yki or Wg may impact the growth potential of cancer cells. The mechanisms by which cancer cells limit the spread of these signals should be elucidated in future studies and may provide molecular insights about how benign and malignant tumors behave.
The biological significance of our findings is validated by other studies that show the formation of context-dependent Yki/YAP-mediated signaling loops as a bona fide mechanism for tumor growth [82,83]. YAP is not only regulated by signaling pathways like Wnt, TGFβ, and notch, but YAP can also collaborate with key cancer pathways to form transcriptional complexes that alter transcriptional programs, specifically in cancer cells [84,85]. At least three different mechanisms have been reported. First, YAP and its cognate transcription factors like TEAD control transcription by binding to promoters of target genes. Second, YAP collaborates with other transcription regulators to alter gene expression, for example, in colon cancer cell lines YAP1, β-catenin, and TBX5 transcriptional complex regulate (TCF- or TEAD-independent) target genes [86]. Third, YAP/TEAD binds distal regulatory elements to regulate the transcription of target genes [87,88,89]. ChIP-seq and deep-sequencing studies in cancer and normal cells showed that several YAP-binding regions also show a consensus motif for AP-1 transcriptional factors, suggesting that YAP/TEAD and AP-1 cooperatively regulate target genes representing a cross-talk between YAP and JNK signaling [88,89].
These findings are especially interesting considering our identification of the tumor-cell-specific Wg–Dronc–JNK–Yki molecular network and the JNK–Yki positive feedback signal amplification loop in the Ras^V12^ scrib^−^ cells. Like YAP, Drosophila Yki is known to regulate transcription by binding to the promoter regions of genes [90]. Similarly, Yki can form transcriptional complexes that cooperatively alter gene expression in cancer cells, for example, Yki and the Ecdysone receptor coactivator Taiman were shown to alter the transcriptional output of Yki-inducible Taiman-dependent genes in cells with hyperactive Yki and neoplastic tumor growth [91]. Based on our data, it is possible that under conditions of oncogenic cooperation, Yki and the Drosophila AP-1 transcription factors may (cooperatively) bind other regulatory regions to drive altered transcriptional output in cancer cells. This conclusion is further strengthened by the finding that dFos, a component of the Drosophila AP-1 (Drosophila Jun and Fos heterodimer) transcription factor, is required for the JNK-mediated invasiveness of Ras^V12^,scrib^−^ tumors [44,66]. In summary, cancer cells may establish specific molecular networks that promote tumor growth and metastasis, and it may be critical to understand these interactions to understand how oncogenic pathways are activated.
5. Conclusions
Our data are significant in light of emerging data from cancer genome studies that revealed that somatic mutations accrued by precancerous cells essentially activate hallmark cancer pathways that converge on a small number of protein complexes and signaling cascades [6]. Our genetic analyses indicate the hierarchy of one such signaling interaction, with Wg acting upstream of Dronc, JNK, and Yki, and in the future, it will be interesting to identify the molecular mechanisms underlying the establishment and regulation of this module. These approaches can also unveil targets for the modulation of the tumor and provide insights into the biomechanics of tumor progression. Our approach of modeling altered signaling interactions in a genetically tractable model is a powerful way to reconstruct key biologically meaningful changes in signaling pathways in cancer cells and provides a functional framework to study the changes in signaling pathway interactions that will generate new insights on mechanisms that promote tumor growth and progression.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Blum A. Wang P. Zenklusen J.C. Snap Shot: TCGA-Analyzed Tumors Cell 201817353010.1016/j.cell.2018.03.05929625059 · doi ↗ · pubmed ↗
- 2Mertins P. Tang L.C. Krug K. Clark D.J. Gritsenko M.A. Chen L. Clauser K.R. Clauss T.R. Shah P. Gillette M.A. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography–mass spectrometry Nat. Protoc.2018131632166110.1038/s 41596-018-0006-929988108 PMC 6211289 · doi ↗ · pubmed ↗
- 3Sanchez-Vega F. Mina M. Armenia J. Chatila W.K. Luna A. La K.C. Dimitriadoy S. Liu D.L. Kantheti H.S. Saghafinia S. Oncogenic Signaling Pathways in The Cancer Genome Atlas Cell 2018173321337.e 1010.1016/j.cell.2018.03.03529625050 PMC 6070353 · doi ↗ · pubmed ↗
- 4Weinstein J.N. Collisson E.A. Mills G.B. Shaw K.R.M. Ozenberger B.A. Ellrott K. Shmulevich I. Sander C. Stuart J.M. The Cancer Genome Atlas Research Network, The Cancer Genome Atlas Pan-Cancer analysis project Nat. Genet.2013451113112010.1038/ng.276424071849 PMC 3919969 · doi ↗ · pubmed ↗
- 5De Vreede G. Gerlach S.U. Bilder D. Epithelial monitoring through ligand-receptor segregation ensures malignant cell elimination Science 202237629730110.1126/science.abl 421335420935 PMC 9197687 · doi ↗ · pubmed ↗
- 6Vogelstein B. Papadopoulos N. Velculescu V.E. Zhou S. Diaz L.A. Kinzler K.W. Cancer Genome Landscapes Science 20133391546155810.1126/science.123512223539594 PMC 3749880 · doi ↗ · pubmed ↗
- 7Torkamani A. Verkhivker G. Schork N.J. Cancer driver mutations in protein kinase genes Cancer Lett.200928111712710.1016/j.canlet.2008.11.00819081671 PMC 2905872 · doi ↗ · pubmed ↗
- 8Pagliarini R.A. Xu T. A Genetic Screen in Drosophila for Metastatic Behavior Science 20033021227123110.1126/science.108847414551319 · doi ↗ · pubmed ↗