Asymmetric Synthesis of Chiral 2-Cyclohexenones with Quaternary Stereocenters via Ene-Reductase Catalyzed Desymmetrization of 2,5-Cyclohexadienones
Michael Friess, Amit Singh Sahrawat, Bianca Kerschbaumer, Silvia Wallner, Ana Torvisco, Roland Fischer, Karl Gruber, Peter Macheroux, Rolf Breinbauer

TL;DR
Scientists developed a new method to efficiently create chiral molecules with complex 3D shapes using enzymes, which is important for drug development.
Contribution
A novel enzymatic method for asymmetric synthesis of chiral 2-cyclohexenones with quaternary stereocenters is introduced.
Findings
Ene-reductases OPR3 and YqjM enable desymmetrizing hydrogenation with high enantioselectivity (>99% ee).
Mechanistic insights were obtained through experimental and theoretical studies.
The chiral enones can undergo diversification reactions while preserving stereochemistry.
Abstract
Stereoselective synthesis of quaternary stereocenters represents a significant challenge in organic chemistry. Herein, we describe the use of ene-reductases OPR3 and YqjM for the efficient asymmetric synthesis of chiral 4,4-disubstituted 2-cyclohexenones via desymmetrizing hydrogenation of prochiral 4,4-disubstituted 2,5-cyclohexadienones. This transformation breaks the symmetry of the cyclohexadienone substrates, generating valuable quaternary stereocenters with high enantioselectivities (ee, up to >99%). The mechanistic causes for the observed high enantioselectivities were investigated both experimentally (stopped-flow kinetics) as well as theoretically (quantum mechanics/molecular mechanics calculations). The synthetic potential of the resulting chiral enones was demonstrated in several diversification reactions in which the stereochemical integrity of the quaternary stereocenter…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Scheme 1
Scheme 1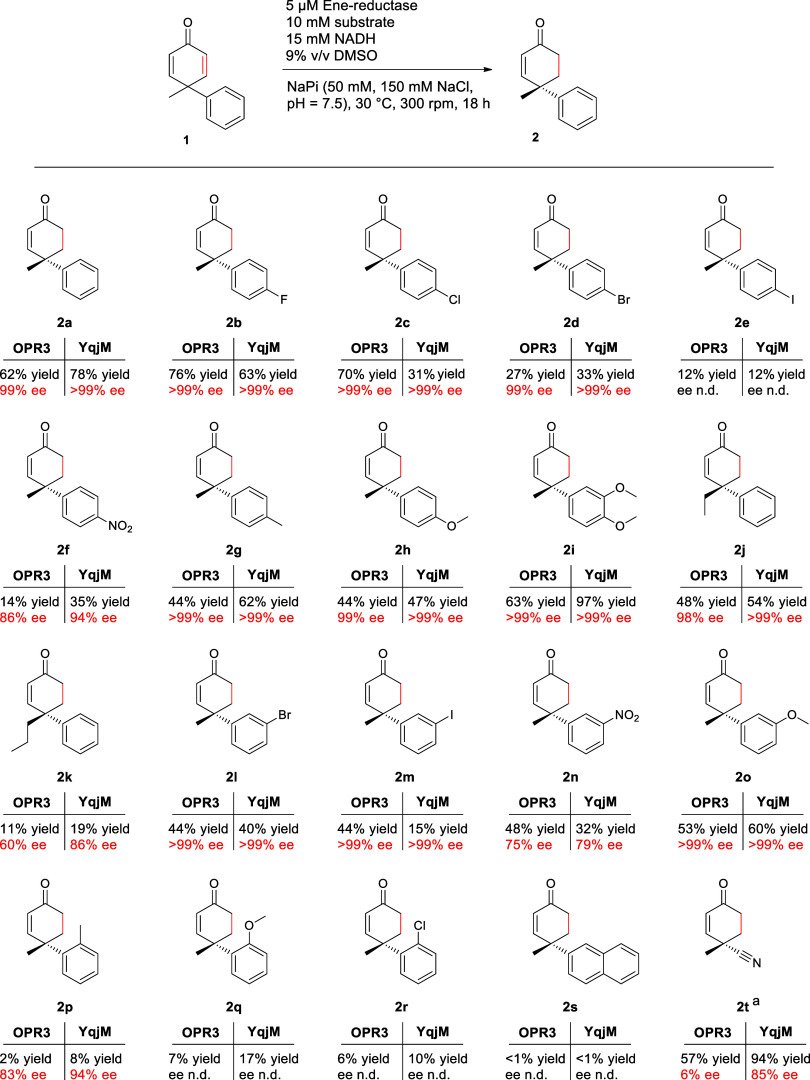 Scheme 2
Scheme 2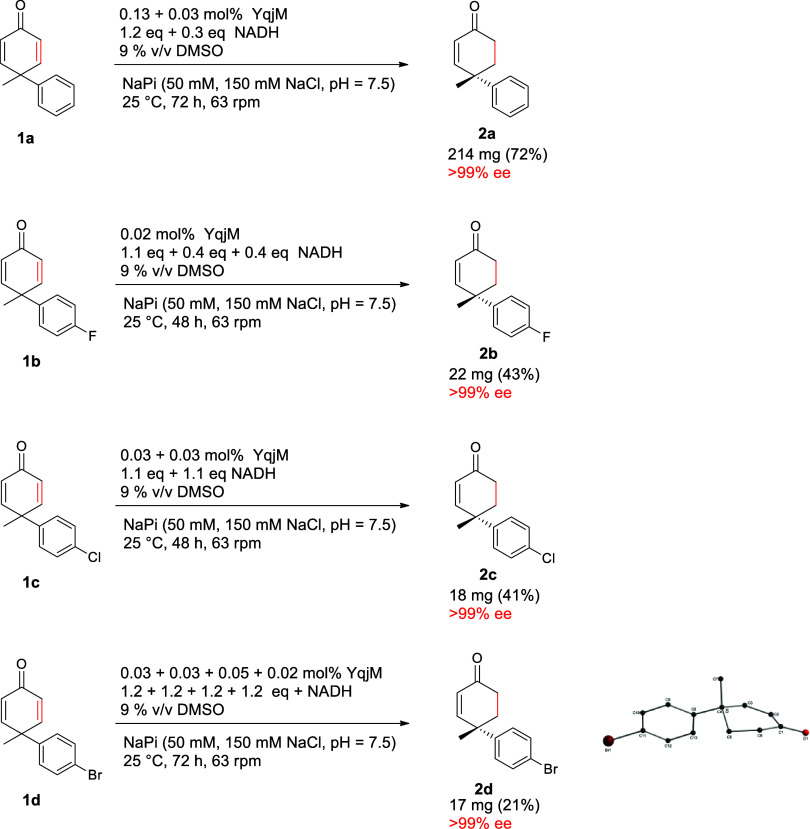 Scheme 3
Scheme 3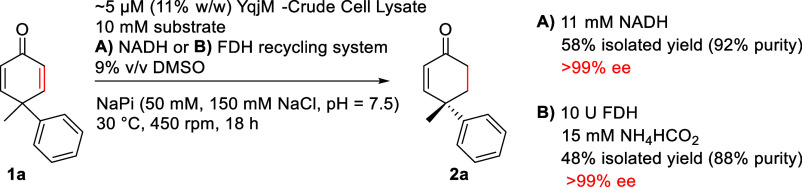 Scheme 4
Scheme 4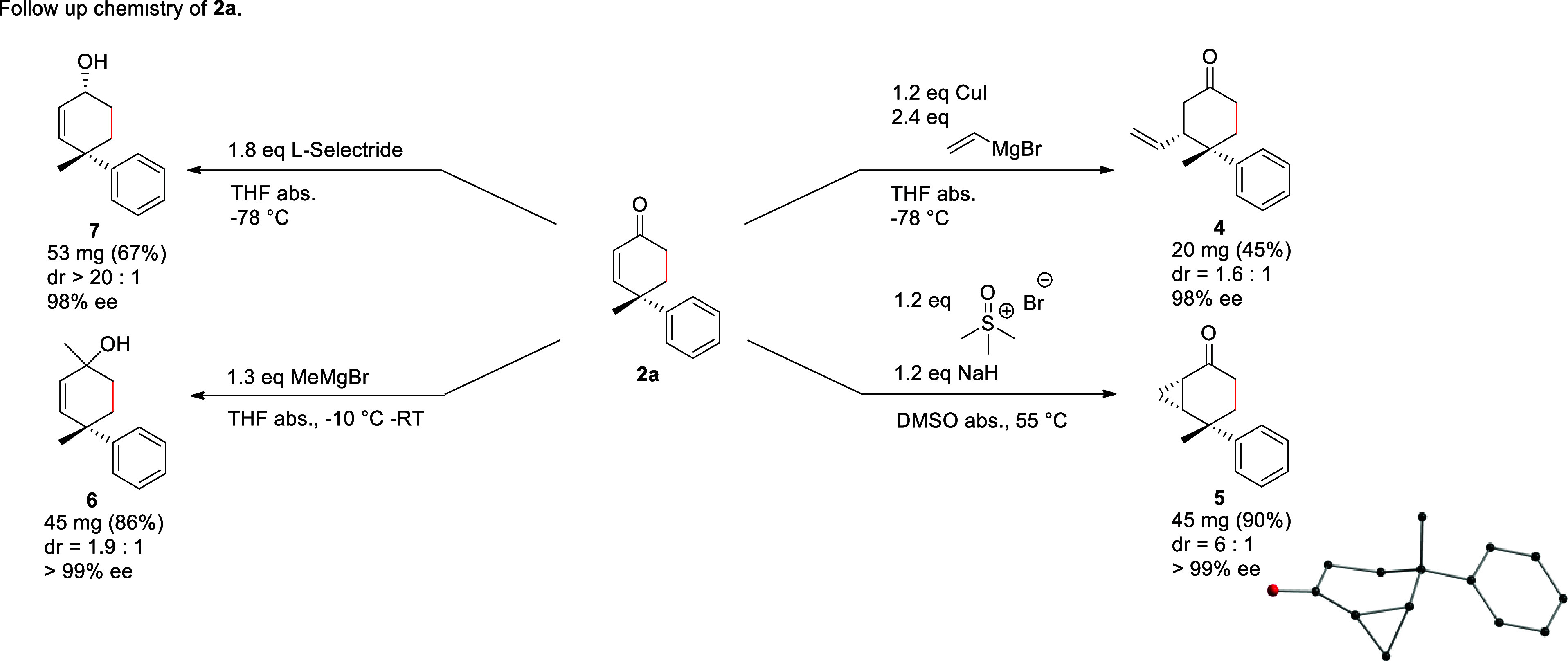 Scheme 5
Scheme 5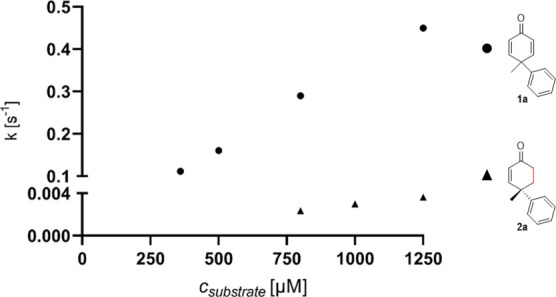 Figure 1
Figure 1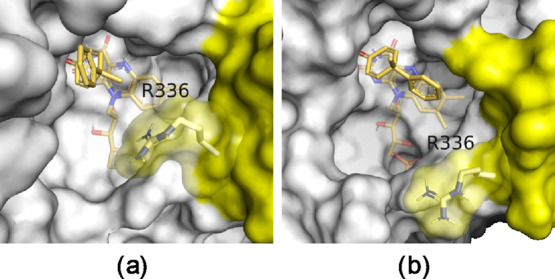 Figure 2
Figure 2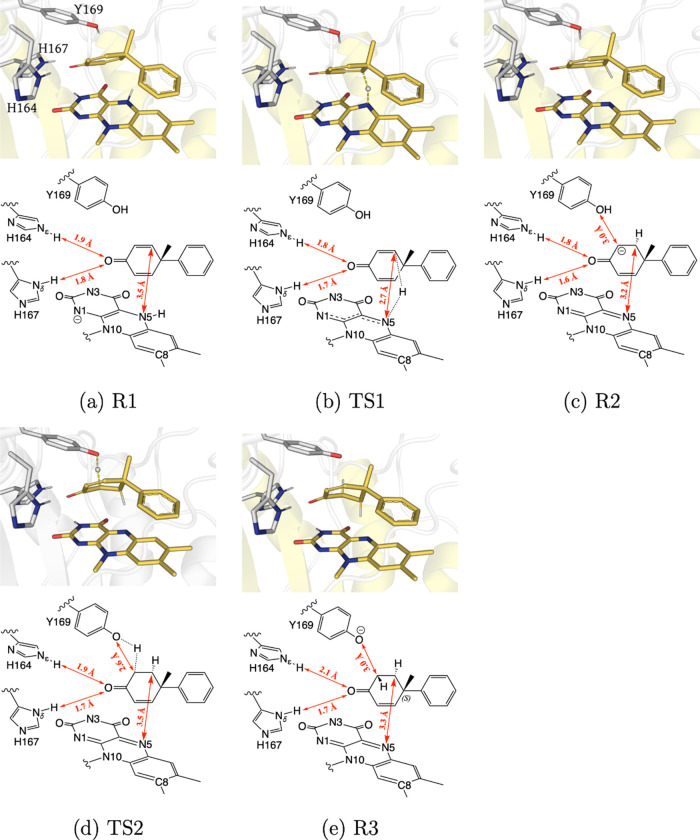 Figure 3
Figure 3| entry | shaking velocity [rpm] | temperature [°C] | time [h] | c-NADH [mM] | DMSO [% v/v] | Yield— |
|---|---|---|---|---|---|---|
| 1 | 300 | 25 | 1 | 15 | 9 | 78 |
| 2 | 400 | 25 | 1 | 15 | 9 | 73 |
| 3 | 500 | 25 | 1 | 15 | 9 | 75 |
| 4 | 300 | 23 | 1 | 15 | 9 | 72 |
| 5 | 300 | 27 | 1 | 15 | 9 | 78 |
| 6 | 300 | 30 | 1 | 15 | 9 | 81 |
| 7 | 300 | 30 | 18 | 15 | 6 | 76 |
| 9 | 300 | 30 | 18 | 20 | 6 | 73 |
| 10 | 300 | 30 | 18 | 20 | 9 | 79 |
- —TU Graz, Internationale Beziehungen und Mobilitätsprogramme10.13039/100008332
- —Karl-Franzens-Universität Graz10.13039/501100009057
- —Austrian Science Fund10.13039/501100002428
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiovascular Health and Risk Factors
Introduction
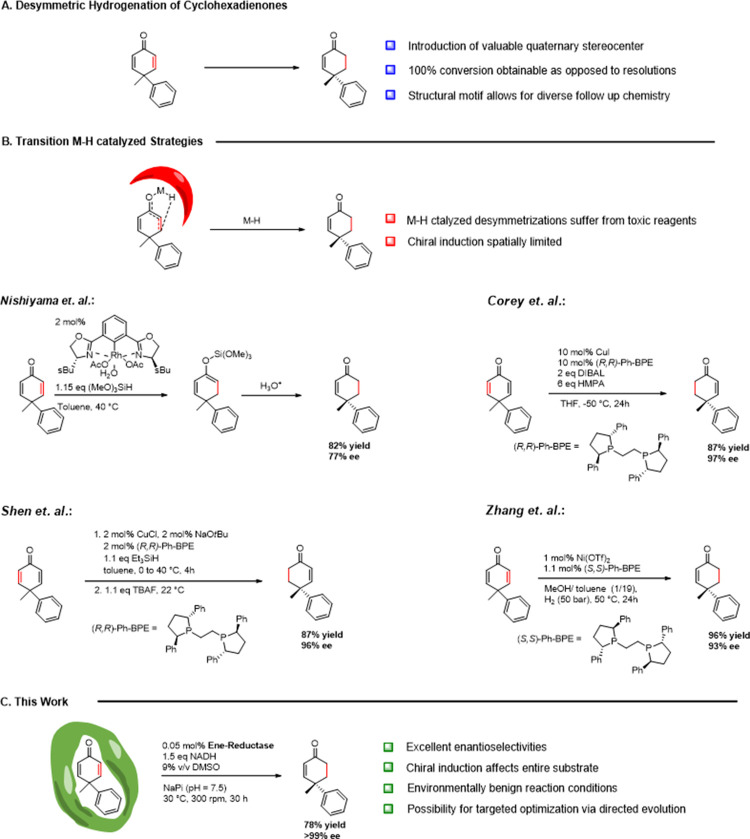
Quaternary all-carbon stereocenters are structural motifs frequently occurring in natural products and pharmaceutically active compounds. Despite their importance, the generation of quaternary stereogenic centers still represents a formidable synthetic challenge.^1−21^ While the stereoselective synthesis of such stereocenters at positions in close vicinity to carbonyl functionalities (α,α-difunctionalized^22−28^ and β,β-difunctionalized carbonyl compounds^29,30^) has been relatively well investigated, the formation of quaternary stereocenters in remote positions to an activating functional group is considerably less explored. For synthesizing carbonyl compounds carrying a quaternary stereocenter in γ-position, desymmetrizing 4,4-disubstituted 2,5-cyclohexadienones has been established as an efficient strategy.^21,31−40^ Such desymmetrization strategies most often rely on intramolecular functionalization of one double bond via the Stetter reaction,^41,42^ Michael addition,^43−45^ Diels–Alder,^46^ or [2 + 3]-cycloaddition^47^ reaction motifs. Furthermore, it was shown that intermolecular Michael addition reactions^48,49^ and Diels–Alder reactions^50^ could be used for cyclohexadienone desymmetrization. These desymmetrization reactions rapidly add complexity to the core cyclohexadienone structure by selectively attaching a substituent to one of the two double bonds. However, if, for certain synthetic applications, no additional functionalization at the α- or β-position is desired, a desymmetrizing hydrogenation of a cyclohexadienone would be ideal for establishing the desired quaternary stereocenter within a 2-cyclohexenone structure, allowing synthetic flexibility to access various synthetic destinations (Scheme 1A). In 2016, Nishiyama reported the first example of such a desymmetrizing hydrogenation using a chiral Rh catalyst with a pincer ligand producing 2-cyclohexenones in up to 77% ee.^51^
Strategies for the Desymmetrizing Hydrogenation of Cyclohexadienones; (A) Principle of Desymmetrizing Hydrogenation; (B) Previous Work with Transition Metal-Catalyzed Reactions; (C) This Work: Employing Ene-Reductases for the Desymmetrizing Hydrogenation of 2,5-Cyclohexadienones under Environmentally Benign Conditions
In an attempt to overcome the dependence on precious Rh metal as part of the catalytic system, Corey reported a Cu-catalyzed desymmetrizing hydrogenation with Ph-BPE as a chiral ligand and DIBAL as a stoichiometric reducing agent (up to 97% ee).^52^ However, superstoichiometric amounts of carcinogenic HMPA were required. These disadvantages could be overcome by Shen and co-workers using the same chiral catalyst but with Et_3_SiH as the stoichiometric reducing agent (up to 96% ee) (Scheme 1B).^53^ Another earth-abundant metal-catalyzed cyclohexadienone desymmetrization was reported by Zhang and co-workers,^54^ in which a Ni/Ph-BPE complex was used for double bond reduction (up to 93% ee) requiring an elevated H_2_ pressure of 50 bar (Scheme 1B).
In contrast to transition-metal catalysis, biocatalysis relies on enzymes as catalysts. Enzymes typically operate under environmentally benign conditions^55^ and can be engineered via well-established methods.^56,57^ Building on our previous work with ene-reductases,^58^ we reasoned that a biocatalytic desymmetrizing hydrogenation of cyclohexadienones could be catalyzed by ene-reductases (Scheme 1C). Ene-reductases from the old yellow enzyme (OYE) family are known to reduce activated double bonds by employing their noncovalently bound flavin cofactor.^59−65^ In contrast to the transition metal-catalyzed hydrogenation reactions described above, in which the stereochemical induction resulted from the coordination of a chiral metal complex to the C=O atom, we were expecting that a biocatalytic desymmetrizing hydrogenation of cyclohexadienones would lead to higher enantioselectivities since this approach would source its chiral induction from the all enclosing nature of an ene-reductase-active site (Scheme 1C). As the stereochemistry-inducing residues are engulfing the substrate in its entirety, we assumed that a potential ene-reductase catalyzed desymmetrizing hydrogenation would be even better suited to generate a quaternary stereocenter in the γ-position—which is spatially quite distant from the cyclohexadienone carbonyl moiety—than it is the case for the aforementioned metal hydride catalyzed approaches.
Results and Discussion
4-Methyl-4-phenyl-cyclohexa-2,5-dienone (1a) was used as a model substrate for the envisioned biocatalytic 2,5-cyclohexadienone desymmetrization process to benchmark its performance in comparison with previously described metal-catalyzed processes. Initial experiments were performed at 25 °C with an enzyme concentration of 5 μM. The employed amount of NADH was held at an equimolar level to avoid overreduction as a potential side reaction. Furthermore, DMSO was used as cosolvent in a concentration of 9% v/v to ensure sufficient solvation of the rather hydrophobic substrate. YqjM wt^66^ and OPR3 wt^67^ were chosen as enzymes as they can be expressed in sufficient quantities and both ene-reductases have been shown to be very stable under various conditions (pH, temperature, solvents), which makes them suitable for preparative biocatalytic processes. We were pleased to see that both tested enzymes delivered the desired enone in high conversion with only minor amounts of fully reduced cyclohexanone being formed. As judged from these initial GC-MS measurements, YqjM appeared to be the more active enzyme in this desymmetrization process. Chiral HPLC showed that cyclohexenone 2a was formed in >99% ee (Table 1, Entry 1). We were delighted that this very first experiment for a biocatalytic hydrogenative desymmetrization had already shown a higher asymmetric induction than all previously reported transitional-metal-catalyzed processes.
Table 1: Optimization of Biocatalytic 2,5-Cyclohexadienone Desymmetrizationa
In order to optimize the conditions of the observed biocatalytic desymmetrization process, parameters like shaking velocity, NADH concentration, and temperature were screened (Table 1). First, it was found that the variation of the shaking velocity had almost no influence on the yield of 2a. Therefore, 300 rpm were used in the following experiments. When varying the temperature, the best results after 1 h were obtained at 30 °C (Entry 6). Increasing the reaction time did not significantly alter the observed 2-cyclohexenone (2a) formation (Entries 8–11), indicating a high kinetic preference for the hydrogenation of the first of the two double bonds of the substrate. When the DMSO concentration was reduced from 9 to 6% v/v, a slightly decreased formation of 2a was observed (Entries 7–10). As an attempt to improve the yield of 2a by promoting the biotransformation with an increased NADH loading of 2 equiv (Entries 9 and 10) did not lead to distinctive improvements, we continued to use 1.5 equiv of NADH and decided to carry out our biotransformations in 18 h reaction time.
We investigated the versatility of our desymmetrization process by testing various 4,4-disubstituted 2,5-cyclohexadienones with OPR3 as well as YqjM as the biocatalyst (Scheme 2).
Substrate Scope of the Ene-Reductase Catalyzed Cyclohexadienone DesymmetrizationReactions were performed in triplicates. Cyclohexenone yields were determined by HPLC at a wavelength of 254 nm. For quantification, external calibrations aided by 1,3,5-tribromobenzene (TBB) as an external standard were used. Substrate-recoveries as determined by HPLC-MS are summarized in the Supporting Information (Table S3). Enantioselectivities were determined by chiral HPLC (CHIRACEL OJ-H): (a) For the biotransformation of 1t, only 1.1 eq NADH were used, and the reaction time was reduced to 3 h. n.d. = not determined.
We observed that the obtained 2-cyclohexenone yields were affected by the size of the substituent in the para-position of the 4-phenyl substituent (Scheme 2). Nonsubstituted substrate 1a was desymmetrized by the better-performing YqjM with an >99% ee in 78% yield, translating to a turnover number (TON) of 1560. 4′-Fluoro-4-phenyl-substituted cyclohexenone (2b) was obtained in yields of 76 and 63% for OPR3 and YqjM, respectively. These yields are similar to those obtained in the desymmetrization of 1a and correspond to TONs of 1520 for the OPR3 and 1260 for the YqjM. The productivity began to drop when substrates with larger halogen atoms at this position were transformed. Notably, the 4′-chloro-substituted substrate 1c showed a remarkable difference depending on the employed enzyme. In this case, OPR3 delivered 2c in a good yield of 70%, while only a 31% yield was observed with YqjM as the biocatalyst. 4′-Bromo- and 4′-iodo-substituted substrates 1d and 1e delivered with the better-performing YqjM enzyme the corresponding 2-cyclohexenones 2d and 2e in yields of 33 and 12%, respectively. In these biotransformations, the mass balance was mostly complemented by the respective unreacted dienone substrates (Supporting Information). Interestingly and much to our delight, the enantioselectivity of the produced products was not affected by increasing the size of the para-halogenide substituent, as 2b–2d were generated in >99% ee for both tested enzymes. 4′-Nitro-4-phenyl-substituted substrate 1f was converted by YqjM to 2f in 35% HPLC-yield but very good enantioselectivity (94% ee). A methyl group in this para-aryl position (1g) was better accepted, producing the corresponding 2-cyclohexenone 2g in 62% yield (>99% ee). 4′-Methoxy-substituted substrate 1h was converted in 44% yield with OPR3 and 47% yield with YqjM with high enantioselectivities for both enzymes (99% for OPR3, >99% ee for YqjM). Interestingly, the sterically more demanding 3′,4′-dimethoxy-4-phenyl substituted substrate 1i was converted to the corresponding 2-cyclohexenone 2i in higher yields than 1h by both investigated ene-reductases (63% for OPR3 and 97% for YqjM) in enantiopure form (>99% ee). This corresponds to a TON of 1940 for the better-performing YqjM and suggests that, in addition to sterics, electronic factors might also influence the substrate-enzyme interactions. The steric influence was also investigated in a series in which we replaced the methyl substituent in standard substrate 1a with ethyl (1j) and propyl (1k) groups. While the transformation of ethyl-substituted substrate 1j with YqjM gave the corresponding enone 2j in 54% yield (>99% ee), the propyl-substituted substrate 1k was converted to enone 2k in only 19% yield (86% ee), exhibiting a strong steric influence. Meta-substituted substrates were quite well tolerated. 3′-Bromo-substituted 2,5-cyclohexadienone 1l delivered the respective enone 2l in 44% yield (>99% ee) with OPR3 and 40% yield (>99% ee) with YqjM. As already observed with para-substituted substrates, an iodo-substituent (1m) also led to the lowest yields within the group of meta-substituted substrates. In contrast, 3′-nitro- (1n) and 3′-methoxy- (1o) substituted substrates were converted with good conversions. Limitations in the reaction were found for *ortho-*substituted substrates 1p-1r, which were converted only in low yields of 2–17%, and naphthyl-substituted substrate 1s was sterically too demanding for the investigated enzymes. In this case, the desired enones could only be found in trace amounts.
In order to test the limits of our reaction, we aimed for the synthesis of 4-cyano-4-methyl-2-cyclohexenone (2t), which has been recently used as an intermediate in the divergent total syntheses of several napelline-type C-20 diterpenoid alkaloid natural products and could be accessed via an asymmetric Diels–Alder reaction with 88% ee.^68^ For the envisioned alternative access using our biocatalytic desymmetrization reaction, the required cyclohexadienone 1t was regarded as a particularly challenging substrate as the methyl and cyano groups are similar in size (Scheme 2). In fact, we found that 1t is much more susceptible to overreduction than the previously described aryl substituted substrates. Fortunately, we could overcome this problem by reducing the amount of utilized cofactor to 1.1 eq NADH. Furthermore, the reaction time was reduced to 3 h for this substrate. Under these conditions, 1t was converted to enone 2t in 57 and 94% yields with OPR3 and YqjM, respectively. With OPR3, almost no stereoinduction was observed (6% ee). However, YqjM delivered nitrile carrying enone 2t in 85% ee. This highly enantioselective conversion of 1t highlights the ability of enzymes to induce chiral induction into substrates with only marginally discriminated substituents, allowing control even of stereogenic centers formed in distant positions to C=O groups required for activation of substrates.
After having established the scope and limitations for our reaction, we wanted to perform our biocatalytic desymmetrization on a preparative scale for several substrates (Scheme 3). For this purpose, the biotransformations were carried out with YqjM as the biocatalyst in Falcon tubes and incubated in an incubation shaker (27 °C, 64 rpm). In the case of substrate 1a, the biotransformation delivered enone 2a in 72% yield (>99% ee). The reaction required the addition of additional enzymes as well as cofactors during the transformation to achieve this yield. Thus, at a preparative scale, YqjM desymmetrization of 1a was achieved with a TON of 450. Para-fluorinated and -chlorinated compounds 2b and 2c could also be isolated in decent yields of 43% (>99% ee) and 41% (>99% ee), respectively. Brominated substrate 1d, which is less soluble in the reaction mixture than 1a–c, turned out to be more challenging to scale up. This substrate required additional enzymes and cofactors throughout the biotransformation. 1d could only be driven to 35% conversion as judged by GC-MS, which resulted in the isolation of 17 mg (21% yield, > 99% ee) of 2d. The absolute configuration of 2d could be assigned via X-ray crystallography. The obtained (S)-configuration of 2d is in accordance with the measured optical rotations of the isolated enone, which are in line with the literature data of this product.^53^
Preparative Scale Ene-Reductase Catalyzed Desymmetrizations
While the use of purified enzymes served us quite well for establishing the scope and limitations of this biocatalytic desymmetrization, larger scale applications would profit from the use of better available enzyme preparations in the form of crude cell lysates. Therefore, we carried out reactions with crude YqjM cell lysate (Scheme 4) (∼5 μM/11% w/w). When this system was used with 11 mM NADH (1.1 equiv), 2a could be isolated in 58% yield [92% purity with the respective cyclohexanone (from overreduction) making up for the remaining 8%].
Crude Cell Lysate-Based Biocatalytic Desymmetrization of 1a with and without a FDH-Cofactor Recycling System
Finally, we showed that our YqjM-crude cell lysate-based desymmetrization of 1a could also be combined with a formate dehydrogenase (FDH)-based cofactor recycling system.^69,70^ For this purpose, we did several screening reactions, which are summarized in the SI (Table S2). Based on this screening, we used 10 U FDH for our 600 μL biotransformations. As terminal reductant 15 mM NH_4_CO_2_H were used. Under these conditions, 2a could be isolated in 48% yield (88% purity; Scheme 4). Due to NAD(P)H already contained in the used crude cell lysate the desymmetrization using FDH-based cofactor recycling did not require any addition of NADH. Furthermore, we were delighted to see that both crude cell lysate-based systems shown in Scheme 4 produced 2a in >99% ee. However, close reaction monitoring should be performed to minimize the overreduction product.
The synthetic value of these generated chiral cyclohexenones can be leveraged by exploiting the enone moiety for the introduction of a diverse array of structurally different substituents. We used several diversification strategies to increase the complexity of compound 2a (Scheme 5). The addition of enone 2a to in situ generated vinyl-cuprate yielded Michael addition product 4 in 45% yield as the 3,4-cis-diastereomer (dr >1.6:1). Similarly, a Corey–Chaykovsky reaction generated cyclopropane 5 in 90% yield (dr 6:1). The preference for the cis-diastereomer can be explained by an axial attack of the trimethylsulfoxonium ylide reagent at the enone half-chair obtaining a chair-conformation as explained by the Fürst–Plattner rule.^71^ The enantiomeric purity of the quaternary stereocenter could be fully retained. The 1,2-reactivity of enone 2a was exemplified for further diversification by Grignard addition with MeMgBr producing alcohol 6 in 86% yield (dr 1.9:1). Similarly, 2a could be reduced with l-selectride to alcohol 7 in 67% yield under nearly full retention of stereoselectivity. Due to the preference of L-selectride to attack in a pseudoequatorial trajectory, 7 was mainly formed as its 1,4-cis-diastereomer (dr >20:1).
Diversification Reactions with Cyclohexenones 2a
The examples described above highlight the synthetic value and potential of the ene-reductase mediated desymmetric hydrogenation of 4,4-substituted 2,5-cyclohexadienones. In order to gain insight into the observed excellent enantioselectivities of this biocatalytic transformation, we performed stopped-flow measurements for dienone 1a and its corresponding desymmetrization product 2a. Both compounds oxidized prereduced YqjM relatively slowly, and no kinetic saturation could be observed in the accessible concentration range (Figure 1). However, dienone 1a turned out to oxidize the prereduced enzyme more than 120 times faster than enone 2a [kobs (at 800 μM of 1a): 0.29 s^–1^; kobs (at 800 μM of 2a): 0.0024 s^–1^; Figure 1]. This finding suggests that the binding of 1a/2a to the active site of YqjM favors the reduction of the pro-S double bond. Apparently, the active site pocket does not allow for a rotation of the quinone moiety in order to place the pro-R double bond near the (reduced) isoalloxazine ring of the flavin cofactor.
Stopped-flow measurements of dienone 1a and enone 2a with YqjM. Measurements were performed against prereduced YqjM under anoxic conditions.
In order to rationalize the stereochemical data obtained with the better-performing YqjM, we conducted a computational analysis employing molecular dynamics (MD) simulations and QM/MM calculations. We modeled the structure of the YqjM complex with 1a using docking and obtained two binding poses of similar energy, in which the carbonyl oxygen atom was hydrogen bonded to the active site histidines H164 and H167 (Figure S7). Binding pose metadynamics was then used to discern the more stable docking pose as starting point for MD simulations. A near-attack conformations (NACs) analysis of the resulting 500 ns MD trajectory revealed a higher preference for the reduction of the pro-S double bond than the pro-R double bond (Figures S7 and S8). YqjM forms a dimer, with residues from both protein chains lining the active site cavity. In the original crystal structure of YqjM in complex with p-nitrophenol,^72^ the side chain of the arginine residue R336 points into the active site, partially blocking access to the flavin cofactor. During the MD simulations, we observed notable conformational changes in R336, opening up the active site and thereby favoring the pro-S binding pose (Figure 2).
Conformational changes in residue R336 of the second protomer observed during the MD simulations. (a) Docked ligand in the crystal structure of the enzyme shows an “in” conformation of R336 restricting the space in the binding site. (b) MD simulations reveal an “out” conformation of R336, enabling more space in the binding site. The two protomers of the YqjM dimer are shown in gray and yellow nontransparent surface representation. Residue R336 is shown as sticks with carbon atoms colored gray and also has a more transparent surface representation, whereas the substrate and flavin are only shown as sticks with their carbon atoms colored yellow.
Quantum mechanics/molecular mechanics (QM/MM) computations were conducted based on a snapshot of the *pro-*S subpopulation resulting from MD simulations. The QM/MM geometry optimized structures corresponding to stationary points along the lowest energy reaction coordinate for the reduction of the pro-S binding pose of 1a are shown in Figure 3. The QM/MM-optimized binding mode of 1a is similar to the one observed for typical OYE substrates like α,β-unsaturated ketones. The electron-withdrawing carbonyl group forms hydrogen bonds with two active site histidines (H164 and H167), resulting in the partial polarization of the C=C double bond and its activation for reduction.^73^ Hydride transfer from the reduced flavin to the C-ß of 1a turned out to be the rate-determining step with a calculated activation energy of 18.1 kcal/mol. The complete energy profile is in accordance with previously reported values^74^ and can be found in Figure S9. In the reactant state (R1) for the hydride transfer step, the distances between the hydride donor atom N5 of the flavin and the pro-S C-ß atom and the pro-R C-ß atom of 1a are 3.5 and 5.1 Å, respectively (Figure 3a). Even though the pro-R C-ß of 1a is further apart from the hydride donor N5 atom, this distance difference does not fully explain the excellent enantioselectivity of our biocatalytic desymmetrization. Another decisive factor for the discrimination between both enantiotopic C-ß positions is the (N10–N5–Cß) angle, which is measured at 101° for the pro-S C-ß atom and 75° for the pro-R C-ß atom. Due to this difference, the hydride can attack the pro-S C-ß via a favorable orthogonal trajectory,^75,76^ whereas it has to settle with a smaller angle of attack to reduce the pro-R C-ß. Another decisive factor for the discrimination between both enantiotopic C-ß positions is the (N10–N5–Cß) angle, which is measured at 101° for the pro-S C-ß atom and 75° for the pro-R C-ß atom. Due to this difference, the hydride can attack the pro-S C-ß via a favorable orthogonal trajectory, whereas it has to settle with a smaller angle of attack to reduce the pro-R C-ß. Similar geometrical constraints were observed in the case of 2a, where the positioning of the double bond over the flavin is unfavorable for an efficient hydride transfer (Figure S10).
(a–e) (Top) QM/MM geometry optimized molecular structures corresponding to stationary points along the lowest energy reaction coordinate for the reduction of 1a. TS1 depicts the transition state for the hydride transfer step, and TS2 displays the transition state for the proton transfer step. The substrate and flavin are shown in yellow sticks, and side chains of active site residues are shown in gray sticks. In TS1 and TS2, the transient hydride and proton are also depicted as a gray ball, respectively. (Bottom) Schematic of each configuration showing key distances (red).
In summary, we have presented the first biocatalytic desymmetrization of 2,5-cyclohexadienones. Due to the strong preference for the pro-S orientation of cyclohexadienone substrates in YqjM and OPR3, cyclohexenones carrying quaternary stereocenters could be generated with excellent enantioselectivities (>99% ee), which surpass previously described methods using enantioselective transition-metal catalysis in respect to both enantioselectivity as well as catalytic efficiency (TON). YqjM was even capable of discriminating between sterically similar methyl- and nitrile-substituents with a high degree of stereoinduction (85% ee) for an intermediate, which can function for the formal total synthesis of napelline-type C-20 diterpenoid alkaloids.^68^ This biocatalytic desymmetric hydrogenation using accessible wild type ene-reductases adds a highly stereoselective and environmentally benign biocatalytic method to the synthetic toolbox, delivering quaternary stereocenters.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Corey E. J.; Guzman-Perez A. The Catalytic Enantioselective Construction of Molecules with Quaternary Carbon Stereocenters. Angew. Chem., Int. Ed. 1998, 37 (4), 388–401. 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE 388>3.0.CO;2-V.29711174 · doi ↗ · pubmed ↗
- 2Zhou F.; Zhu L.; Pan B.-W.; Shi Y.; Liu Y.-L.; Zhou J. Catalytic enantioselective construction of vicinal quaternary carbon stereocenters. Chem. Sci. 2020, 11 (35), 9341–9365. 10.1039/D 0SC 03249 B.34094201 PMC 8162142 · doi ↗ · pubmed ↗
- 3Pierrot D.; Marek I. Synthesis of Enantioenriched Vicinal Tertiary and Quaternary Carbon Stereogenic Centers within an Acyclic Chain. Angew. Chem., Int. Ed. 2020, 59 (1), 36–49. 10.1002/anie.201903188.31081180 · doi ↗ · pubmed ↗
- 4Feng J.; Holmes M.; Krische M. J. Acyclic Quaternary Carbon Stereocenters via Enantioselective Transition Metal Catalysis. Chem. Rev. 2017, 117 (19), 12564–12580. 10.1021/acs.chemrev.7b 00385.28910092 PMC 5651685 · doi ↗ · pubmed ↗
- 5Büschleb M.; Dorich S.; Hanessian S.; Tao D.; Schenthal K. B.; Overman L. E. Synthetic Strategies toward Natural Products Containing Contiguous Stereogenic Quaternary Carbon Atoms. Angew. Chem., Int. Ed. 2016, 55 (13), 4156–4186. 10.1002/anie.201507549.PMC 486501626836448 · doi ↗ · pubmed ↗
- 6Liu Y.; Han S.-J.; Liu W.-B.; Stoltz B. M. Catalytic enantioselective construction of quaternary stereocenters: assembly of key building blocks for the synthesis of biologically active molecules. Acc. Chem. Res. 2015, 48 (3), 740–751. 10.1021/ar 5004658.25715056 PMC 6410712 · doi ↗ · pubmed ↗
- 7Long R.; Huang J.; Gong J.; Yang Z. Direct construction of vicinal all-carbon quaternary stereocenters in natural product synthesis. Nat. Prod. Rep. 2015, 32 (11), 1584–1601. 10.1039/C 5NP 00046 G.26334685 · doi ↗ · pubmed ↗
- 8Wang B.; Tu Y. Q. Stereoselective construction of quaternary carbon stereocenters via a semipinacol rearrangement strategy. Acc. Chem. Res. 2011, 44 (11), 1207–1222. 10.1021/ar 200082 p.21728380 · doi ↗ · pubmed ↗