Heterogeneous genetic patterns in bilateral perisylvian polymicrogyria: insights from a Finnish family cohort
Irma Järvelä, Ritva Paetau, Yasmin Rajendran, Anushree Acharya, Thashi Bharadwaj, Suzanne M Leal, Anna-Elina Lehesjoki, Maarit Palomäki, Isabelle Schrauwen

TL;DR
This study identifies new genetic causes of a brain development disorder called bilateral perisylvian polymicrogyria in a Finnish family group.
Contribution
The study reports the first association of NUS1 and DDX23 with bilateral perisylvian polymicrogyria.
Findings
Pathogenic or likely pathogenic variants were found in 24% of families.
Variants were identified in five genes, including two (NUS1 and DDX23) previously unlinked to the condition.
The findings confirm the genetic heterogeneity of bilateral perisylvian polymicrogyria.
Abstract
Bilateral perisylvian polymicrogyria is the most common form of regional polymicrogyria within malformations of cortical development, constituting 20% of all malformations of cortical development. Bilateral perisylvian polymicrogyria is characterized by an excessive folding of the cerebral cortex and abnormal cortical layering. Notable clinical features include upper motoneuron dysfunction, dysarthria and asymmetric quadriparesis. Cognitive impairment and epilepsy are frequently observed. To identify genetic variants underlying bilateral perisylvian polymicrogyria in Finland, we examined 21 families using standard exome sequencing, complemented by optical genome mapping and/or deep exome sequencing. Pathogenic or likely pathogenic variants were identified in 5/21 (24%) of families, of which all were confirmed as de novo. These variants were identified in five genes, i.e. DDX23, NUS1,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
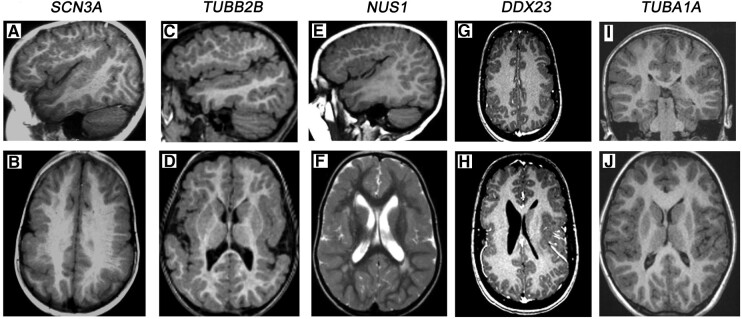 Figure 1
Figure 1| Patient | Sex | Age | Gene | Variant | Inheritance | Severity of ID or DD | MRI/CT | Other features | ACMG | Evidence |
|---|---|---|---|---|---|---|---|---|---|---|
| FINLIS2 | M | 18 |
|
| Mild | BPP | Epilepsy | P | PS2, PM2, PP2, PS1, PS3 | |
| FINLIS4 | F | 22 |
|
| Mild | BPP | Epilepsy | P | PS2, PP2, PM2, PS1_sup, PM1 | |
| FINLIS14 | F | 14 |
|
| Mild | BPP | Hearing impairment | LP | PS2, PM2 | |
| FINLIS18 | F | 19 |
|
| ND | BPP | Rhabdomyosarcoma | LP | PS2, PM2, PP2 | |
| FINLIS22 | F | 11 |
|
| Moderate | Insular cortex | Spastic diplegia | P | PS2, PP3, PM2, PM1, PP2, PM5 | |
|
| ||||||||||
| FINLIS1 | M | 34 |
| AD | Borderline | BPP | Wolfram-like syndrome | LP | PP1_sup, PP3, PM2, PS1 | |
- —Helsinki University Central Hospital10.13039/100008376
- —National Institutes of Health10.13039/100000002
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and Clinical Aspects of Sex Determination and Chromosomal Abnormalities · Connective tissue disorders research · Preterm Birth and Chorioamnionitis
Introduction
Malformations of cortical development (MCD) are rare congenital anomalies of the cerebral cortex falling under neuronal migration defects.^1^ MCDs are classified into three major groups: malformations caused by abnormal cell proliferation, misdirected neuronal migration, or aberrant post-migrational cortical organization and connectivity.^1,2^ Polymicrogyria (PMG) is the most common form accounting for 20% of all MCDs.^3^ PMG is characterized by an overfolding of the cerebral cortex and abnormal cortical layering, which is diagnosed by brain imaging such as MRI. Both genetic (non-syndromic and syndromic forms) and non-genetic causes have been reported for PMG.^4^ Currently, ∼50 genes have been identified as responsible for PMGs, accounting for ∼20–32% of the cases.^4-8^
Bilateral perisylvian polymicrogyria (BPP) is the most common type of regional PMG.^9^ The major clinical features include upper motor neuron dysfunction, dysarthria and asymmetric quadriparesis. Cognitive impairment and different types of epilepsy have also been described. The clinical features in PMG, and even in BPP, can be quite variable and heterogeneous.
For BPP, Mirzaa et al.^5^ identified a recurrent mosaic variant (p.Gly373Arg) in PIK3R2. We previously discovered a pathogenic variant in SCN3A that underlies a unique neurodevelopmental channelopathy in a Finnish BPP cohort.^8^ In this study, we used a stepwise approach of standard exome sequencing (ES), followed by deep ES sequencing of buccal-derived DNA (ectoderm lineage) and/or optical mapping (OGM) to find genetic causes for 21 families in a Finnish BPP cohort.
Materials and methods
Study approval
Written informed consent was obtained from the parents and the patients according to the Declaration of Helsinki. Written informed consent was obtained to publish photographs of the patients. The study was approved by the ethics committees of the Hospital District of Helsinki and Uusimaa (HUS/2532/2017) and the Institutional Review Boards of Columbia University (IRB-AAAS3433).
Family ascertainment and phenotype analysis
A total of 21 Finnish families with 22 cases of BPP detected by MRI were enrolled in the study. The samples were collected between 1998 and 2023 and included 18 trios, 2 duos and 1 singleton case. Affected individuals were clinically evaluated by one of the authors (R.P.). None of the individuals had extrinsic non-genetic causes, including vascular, hypoxic insults or congenital cytomegalovirus infection. Peripheral blood samples were collected from study participants that included both affected and unaffected family members. Genomic DNA was isolated following standard procedures. Families with inconclusive/negative exome results were studied further via deep buccal sequencing and/or OGM. Unfortunately, we were unable to collect a buccal sample and/or an additional blood sample for certain negative/inconclusive families (Supplementary Table 1). Ultra-high-molecular-weight (UHMW) DNA from frozen peripheral blood samples of eight individuals was extracted using the Prep SP Blood and Cell Culture DNA Isolation Kit (Bionano Genomics, San Diego, CA, USA). Buccal epithelial cells were collected from nine affected individuals. Buccal DNA was isolated using the prepIT L2P extraction kit (DNA Genotek Inc., Ottawa, ON, Canada).
ES and bioinformatic analysis
Exomic libraries were prepared using the SureSelect Human All Exon V6 kit (Agilent Technologies, Santa Clara, CA, USA). Paired-end sequencing was performed on a NovaSeq6000 instrument (Illumina Inc., San Diego, CA, USA), targeting 50× for standard ES (using blood-derived DNA) and 200× for deep ES (using buccal-derived DNA). Reads were aligned to the human genome (hg38) using Burrows–Wheeler alignment-maximal exact match (MEM),^10^ and single nucleotide variants (SNVs) and small insertion/deletions (indels) were called following the Genome Analysis Toolkit best practices in variant calling.^11^ Copy number variants (CNVs) were called using Copy Number Inference from Exome Reads^12^ (PMID: 22585873).
Rare variants following several inheritance models (e.g. de novo, autosomal recessive, autosomal dominant and X-linked) with a predicted effect on protein function or pre-mRNA splicing were retained. We also performed an additional analysis to detect variants present only in the ectodermal lineage (buccal). In this case, standard and deep exome sequences of the same cases were called jointly, and variants present in the standard exome sequence data were excluded.
Sanger sequencing was performed using an ABI3130XL Genetic Analyzer to verify candidate SNV and InDel variants and to examine segregation among the family members that were not exome-sequenced. The classification of variants is based on the American College of Medical Genetics and Genomics recommendations.^13^
Optical genome mapping and bioinformatic analysis
UHMW DNA labelling was processed with the Bionano Prep DLS DNA Labeling kit (Bionano Genomics). Labelled DNA was loaded on a Saphyr chip and run on a Saphyr instrument (Bionano Genomics), targeting ∼200× coverage. A de novo genome map assembly was performed using Bionano Solve™, followed by structural variant calling. Data were analysed with Bionano Access™ and was further annotated with annotSV^14^ and biomaRt.^15^ Rare variants, based on their frequency in the Database of Genomic Variants, dbVar (NCBI), gnomAD and 279 controls from Bionano Genomics, were prioritized.
Results
We found novel pathogenic (P)/likely pathogenic (LP) variants in five genes known to underlie a neurodevelopmental disorder in 21 (24%) families with BPP using ES (Table 1; Supplementary Materials). All of them were de novo. Their brain MRI images are presented in Fig. 1. Detailed clinical information is given in Supplementary Materials.
Brain MR images of patients with pathogenic or likely pathogenic gene variants. The subject with an SCN3A variant shows BPP and thick grey matter over the frontal, parietal and perisylvian cortexes (A, B). The subject with a TUBB2B variant has bilateral fronto-parietal, perisylvian and insular polymicrogyria (C, D). The subject with an NUS1 variant has a similar pattern of distribution of polymicrogyria as seen with SCN3A and TUBB2B but additional white matter changes around the posterior horn of the lateral ventricles (E, F). The subject with a DDX23 variant has asymmetric patches of frontal, parietal and insular/perisylvian polymicrogyria (G, H). The polymicrogyric cortex of the subject with a TUBA1A variant is limited to the opercular and insular cortexes (I, J). SCN3A, the sodium voltage–gated channel alpha subunit 3; TUBB2B, tubulin 2B; NUS1, dehydrodolichyl diphosphate synthase subunit; DDX23, DEAD-box RNA helicase 23 gene; TUBA1A, tubulin alpha-1A.
In FINLIS2-3, a de novo variant p.(Arg1621Gln) in the sodium voltage–gated channel alpha subunit 3 (SCN3A) gene (OMIM # 617935) was identified. He presented with severe oral motor dysfunction and autonomic seizures from infancy. At 15 years, he experienced tonic–clonic seizures, but these were ameliorated with valproate and oxcarbazepine treatment. He has mild intellectual disability (ID). A brain MRI identified BPP.
FINLIS4-3 had a de novo missense variant p.(Pro259Leu) in tubulin 2B (TUBB2B) (OMIM # 610031). Her phenotype was characterized by mild right-dominant tetraplegia, severe dysarthria, nocturnal epileptic myoclonus, mild ID and bilateral perisylvian and perirolandic PMG.
FINLIS14-3 had a de novo missense p.(Asp206Tyr) variant in the dehydrodolichyl diphosphate synthase subunit (NUS1; OMIM # 617831). The phenotype was characterized by mild right-hand paresis, severe oral-motor and oral sensory problems, severe congenital sensorineural deafness (diagnosed at 2 years of age) and mild ID. She has no epilepsy or dysmorphism. Her social skills are good but challenged by anxiety in daily activities. Her MRI displayed BPP and bilateral white matter changes around the posterior horns of the lateral ventricles.
FINLIS18-3 had a de novo missense variant p.(Arg813Cys) in the DEAD-box RNA helicase 23 gene (DDX23; OMIM # 612172). The phenotype was severe, including problems of swallowing with aspiration that led to tube feeding and permanent tracheostomy as a newborn. Permanent gastrostomy for feeding was inserted at 2.5 years of age. Due to the hypoplastic lower jaw, the tongue and the epiglottis and the paralysed tongue and pharyngeal muscles, she could not swallow. She learnt to walk from 3 years of age but never learnt to produce any vocalization. At 7 years of age, her neuropsychological profile showed normal performance. A brain MRI at ages 4 and 8 showed BPP. At 16 years of age, an aggressive rhabdomyosarcoma appeared in the left cheek, resistant to radiation and chemotherapies that invaded large areas of the neck and extra-cranial structures, and finally, the left hemisphere. She passed away at 19 years of age.
FINLIS22-3 has a de novo missense variant in the N-terminal region in Exon 3 (p.Asn101Lys) of tubulin alpha-1A (TUBA1A), a well-known gene for MCDs.^16^ Her oral motor function is close to normal, whilst spastic diplegia and moderate ID dominate the phenotype. A brain MRI demonstrated that BPP is mainly located in the insular cortex.
The underlying cause of BPP was not identified for the proband in FINLIS1. However, in addition to BPP, the affected son and his unaffected mother have hearing impairment. We identified a previously reported^17^ missense variant p.(Ser308Cys) in Wolframin ER Transmembrane Glycoprotein (WFS1) underlying autosomal-dominant Wolfram-like syndrome (#OMIM 614296) in both the mother and the son. This agrees with the segregation of autosomal-dominant hearing impairment in this family (data not available) but does not explain BPP.
ES revealed variants of unknown significance in several families (Supplementary Table 2; Supplementary Materials). Deep ES of buccal epithelial DNA was utilized in nine inconclusive/negative cases, where a sample could be obtained. No disease-causing variants were identified (Supplementary Tables 1 and 2). Eight subjects, with available frozen blood samples, whose ES did not reveal a relevant candidate gene, were analysed using OGM, but no variants were identified (Supplementary Table 1). In addition, no CNVs were found through ES.
Discussion
We applied standard ES, followed by deep ES of buccal samples and/or OGM to identify the genetic causes of BPP. We found pathogenic or likely pathogenic variants in 24% of the analysed families, all of which were de novo. The results are similar to those of previous studies.^4,7^ In addition to identifying variants in well-known BPP genes, i.e. TUBA1A, TUBB2B and SCN3A,^6,8^ we propose DDX23, a member of the RNA-helicase gene family,^18^ and NUS1^19^ as novel genes underlying BPP, expanding their phenotypic spectrum.
Interestingly, Zaman et al.^20^ reported the same variant p.(Arg1621Gln) in SCN3A identified by us in a patient with severe ID, and normal EEG and a different variant in the same codon p.(Arg1621Gly), in a patient with severe ID and pseudobulbar palsy. We previously reported a different variant p.(Phe1759Tyr) in SCN3A in a large Finnish family with BPP.^8^ The cognition of the affected individuals ranged from mild ID to borderline intelligence.^8^
TUBA1A and TUBB2B belong to tubulinopathies associated with a wide range of MCDs.^17,21,22^ Among individuals with PMG, including the most common perisylvian subtype, TUBA1A variants have been found in 3.1% of all patients with PMG and in 10% of PMG with complex cerebral malformations.^17^ Pathogenic TUBB2B variants have been estimated to be involved in 2.3% of MCDs.^23^ The clinical phenotypes caused by the variants of TUBA1A can vary considerably; most affected patients have congenital microcephaly and severe neurodevelopmental delay with di/tetraplegia.^1^ FINLIS22-3 had BPP mainly located in the insular cortex. She had normocephaly, a relatively mild upper motor neuron dysfunction and no epilepsy. TUBA1A variants were found as the most common cause of microlissencephalies in foetuses.^22^ The missense variant p.(Asn101Lys) in TUBA1A in FINLIS22-3 was located in the same codon as previously described in a foetus with p.(Asn101Ser) that showed a poorly differentiated cortical plate^22^ and in a person with microlissencephaly.^21^ FINLIS4-3 with a pathogenic missense variant p.(Pro259Leu) in TUBB2B had bilateral perisylvian and perirolandic PMG. Previously, TUBB2B variants have been reported to underlie more extensive brain malformations and severe phenotypes.^24^ Of note, patients with tubulinopathy in our study had a normal corpus callosum, brainstem and cerebellum. Our results further strengthen the role of tubulin genes in BPP and suggest further heterogeneity both in clinical features and in MRI findings.
We propose phenotypic expansions of two known disease genes, NUS1 and DDX23, to include BPP. NUS1 variants are associated with a wide spectrum of phenotypes.^25^ In addition to the characteristic features of BPP (mild right-hand paresis, severe oral-motor problems and oral sensory problems), a patient in this study had severe congenital sensorineural deafness. In her ES, we did not find a separate variant in a gene that would explain her deafness. However, hearing impairment is seen in patients with autosomal recessive NUS1 (a congenital disorder of glycosylation; OMIM # 617082).^26^
DDX23 belongs to the DExD/H-box RNA helicase genes that have been associated with multi-organ diseases, neurodevelopmental disorders, brain malformations and several types of cancer.^18,27,28^ DDX23 (OMIM #612172) has recently been reported to be associated with a novel syndromic neurodevelopmental disorder.^29^ The phenotype includes developmental delay, muscle tone abnormalities and dysmorphic features. Brain abnormalities, including decreased white matter, corpus callosum abnormalities and grey matter heterotopia, have been noted in some patients.^29,30^ The variant p.(Arg813Cys) is located near the C-terminal RecA-like domain at the 3′ end of the DDX23 gene at a highly conserved amino acid (GERP_RS_: 5.97) and may explain the severe phenotype of FINLIS18-3. Yin et al.^28^ have reported that DDX23 has a role in promoting the malignancy of gliomas in the brain. It is intriguing to speculate whether DDX23 was associated with both BPP and rhabdomyosarcoma in the young female FINLIS18-3 in this study. Interestingly, PMG has also been reported in patients with pathogenic variants in another helicase genes DHX37^18^ and DDX3X.^31^
Finally, despite incorporating various genomic techniques alongside standard-coverage ES, the diagnostic yield of BPP in this investigation aligns closely with findings from prior studies. However, it remains notably lower than diagnostic rates for other congenital brain malformations, such as lissencephaly, in which the diagnostic rate is ∼80%.^32^ This discrepancy may be attributed to an increase in somatic variants that are exclusively present in a few or single tissues. In addition, the variant may have a very low-level mosaic in the tissues studied here (blood/buccal) and therefore may need a deeper analysis than 200×. To enhance the identification of causal variants in these disorders, there is a pressing need for the adoption of more sophisticated, preferably minimally invasive sampling techniques and sequencing methodologies.
In summary, our data suggest an increased incidence of phenotypic and genotypic heterogeneity of BPP and call for the use of new methods to elucidate the biological background of BPP.
Supplementary Material
fcae142_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. A developmental and genetic classification for malformations of cortical development: Update 2012. Brain. 2012;135:1348–1369.22427329 10.1093/brain/aws 019PMC 3338922 · doi ↗ · pubmed ↗
- 2Guerrini R, Dobyns WB. Malformations of cortical development: Clinical features and genetic causes. Lancet Neurol. 2014;13:710–726.24932993 10.1016/S 1474-4422(14)70040-7PMC 5548104 · doi ↗ · pubmed ↗
- 3Leventer RJ, Phelan EM, Coleman LT, Kean MJ, Jackson GD, Harvey AS. Clinical and imaging features of cortical malformations in childhood. Neurology. 1999;53:715–722.10489031 10.1212/wnl.53.4.715 · doi ↗ · pubmed ↗
- 4Stutterd CA, Brock S, Stouffs K, et al Genetic heterogeneity of polymicrogyria: Study of 123 patients using deep sequencing. Brain Commun. 2020;3:fcaa 221.10.1093/braincomms/fcaa 221PMC 787824833604570 · doi ↗ · pubmed ↗
- 5Mirzaa G, Conti V, Timms AE, et al Characterization of mutations of the phosphoinositide-3-kinase regulatory subunit, PIK 3R 2, in perisylvian polymicrogyria: A next generation sequencing study. Lancet Neurol. 2015;14(12):1182–1195.26520804 10.1016/S 1474-4422(15)00278-1PMC 4672724 · doi ↗ · pubmed ↗
- 6Parrini E, Conti V, Dobyns WB, Guerrini R. Genetic basis of brain malformations. Mol Syndromol. 2016;7:220–233.27781032 10.1159/000448639 PMC 5073505 · doi ↗ · pubmed ↗
- 7Akula SK, Chen AY, Neil JE, et al Exome sequencing and the identification of new genes and shared mechanisms in polymicrogyria. JAMA Neurol. 2023;80(9):980–988.37486637 10.1001/jamaneurol.2023.2363 PMC 10366952 · doi ↗ · pubmed ↗
- 8Smith RS, Kenny CJ, Ganesh V, et al Sodium channel SCN 3A (Na V 1.3) regulation of human cerebral cortical folding and oral motor development. Neuron. 2018;99(5):905–913.e 7.30146301 10.1016/j.neuron.2018.07.052PMC 6226006 · doi ↗ · pubmed ↗