A Case of Marfan Syndrome With Congenital Hip Dysplasia and Spine Abnormality
Siddhartha Yadao, Kartik Bansal, Shrutika M Mungal, Avni Gakkhar, Ashok M Mehendale

TL;DR
This case report describes an 11-year-old boy with Marfan syndrome who shows unusual musculoskeletal symptoms but no heart issues.
Contribution
The novelty lies in highlighting atypical clinical features of Marfan syndrome without cardiovascular involvement.
Findings
The patient exhibited classical signs like wrist/thumb signs and pectus carinatum, but lacked cardiovascular abnormalities.
Management included beta-blockers, orthopedic care, and ophthalmologic interventions.
The case emphasizes the need for multidisciplinary care and awareness of non-cardiac presentations.
Abstract
Marfan syndrome, a hereditary disorder of connective tissue marked by FBN1 gene mutations, presents a clinical tapestry requiring a multidisciplinary approach for optimal management. This case report details the presentation of an 11-year-old male exhibiting musculoskeletal deformities, notably an abnormally curved spine and congenital hip dysplasia, indicative of Marfan syndrome. The absence of cardiovascular abnormalities and family history challenges the diagnostic process. Clinical evaluation revealed classical signs, including positive wrist and thumb signs, pectus carinatum, a loose skin fold, and scapular winging. Laboratory investigations, including imaging studies, confirmed the diagnosis. The patient’s management involves a multifaceted strategy, addressing cardiovascular risks through beta-blockers and potential surgical interventions, orthopedic measures for musculoskeletal…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
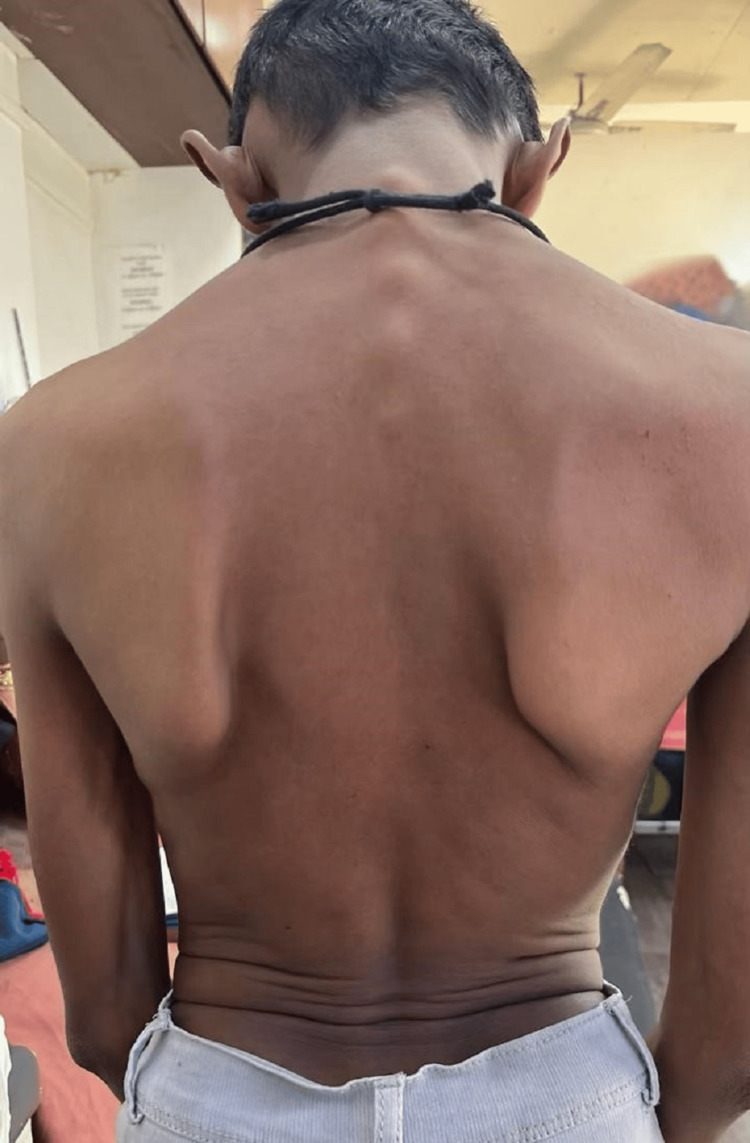 Figure 1
Figure 1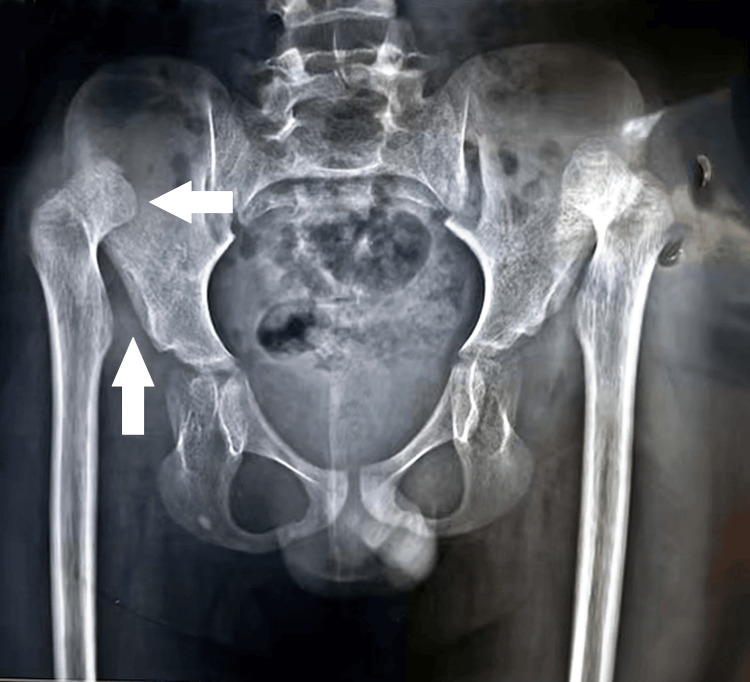 Figure 2
Figure 2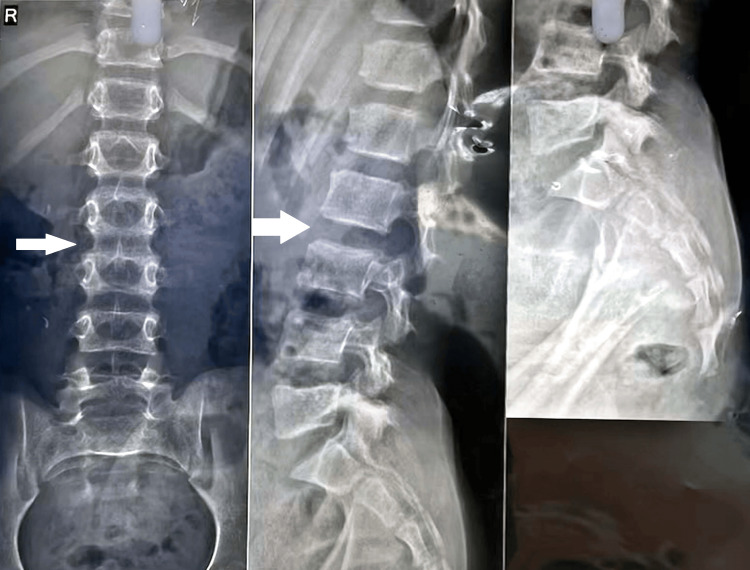 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsConnective tissue disorders research · Shoulder Injury and Treatment · Cardiac Valve Diseases and Treatments
Introduction
Marfan syndrome is a multisystemic, hereditary disorder characterized by abnormalities in connective tissue, predominantly affecting the cardiovascular, skeletal, and ocular systems. First described by the French physician Antoine Marfan in 1896, this syndrome has since been recognized as one of the most common inherited connective tissue disorders, with an estimated incidence of 1 in 5,000 individuals worldwide. While the exact prevalence may vary among different populations and regions, Marfan syndrome remains a significant clinical entity with profound implications for affected individuals and their families [1]. The underlying pathophysiology of Marfan syndrome stems from mutations in the fibrillin-1 gene (FBN1), located on chromosome 15q21.1, which encodes for fibrillin-1, a significant component of microfibrils found in connective tissue [2]. These mutations lead to structural abnormalities in the extracellular matrix, resulting in systemic manifestations characteristic of the syndrome. Furthermore, approximately 25-30% of cases are due to de novo mutations with no family history of the disorder, underscoring the importance of recognizing sporadic cases in clinical practice [3]. Clinically, Marfan syndrome presents with a broad spectrum of features, varying in severity and age of onset [4]. Cardinal manifestations typically involve the musculoskeletal, cardiovascular, and ocular systems, although other organ systems may be affected to varying degrees. Skeletal abnormalities, such as tall stature, arachnodactyly (long, slender fingers), joint hypermobility, and spinal deformities, are often among the earliest signs observed in affected individuals. These skeletal manifestations can result in functional limitations and orthopedic complications, contributing to the morbidity associated with the syndrome [5]. Cardiovascular involvement represents a significant concern in Marfan syndrome, with a predisposition to aortic root dilation, aortic dissection, and valvular abnormalities. Aortic root dilation, in particular, poses a substantial risk of life-threatening complications, emphasizing the importance of regular cardiac surveillance and timely interventions to mitigate the risk of aortic dissection [1,2]. Additionally, ocular manifestations, including lens dislocation, myopia, and retinal detachment, further contribute to the clinical complexity of Marfan syndrome, necessitating comprehensive ophthalmologic evaluation and management [6]. Despite the advancements in our understanding of the genetic basis and clinical manifestations of Marfan syndrome, challenges remain in achieving early diagnosis and implementing appropriate management strategies [3]. This particular case requires timely recognition and intervention, especially by orthopedics, spine specialists, physiotherapists, and psychiatrists, as well as genetic counseling [4-6].
Case presentation
An 11-year-old male presented with a chief complaint of difficulty walking and bearing weight since childhood, associated with an abnormally curved spine. The patient’s medical history revealed no palpitations, autonomic abnormalities, pain, or respiratory distress. Additionally, there was no familial history of similar symptoms or known genetic disorders. On physical examination, the patient exhibited a tall, thin build with musculoskeletal deformities, including positive wrist and thumb signs, pectus carinatum, a loose skin fold, and winging of the scapula (Figure 1). The general examination was otherwise normal despite these findings, and no cardiovascular abnormalities were observed. The respiratory examination demonstrated pectus carinatum. Laboratory investigations, including a complete blood count, blood urea and electrolytes, liver and kidney function tests, growth hormone, and troponin, were within normal limits. However, a chest X-ray revealed an increased cardiothoracic ratio, and an X-ray of the pelvis indicated congenital hip dysplasia (Figure 2). An X-ray of the spine demonstrated a prominent vertebral process, increased disc space, and winging of the scapula (Figure 3). Based on the clinical features and imaging findings, the patient was diagnosed with Marfan syndrome. While the absence of cardiovascular abnormalities at this stage was noted, regular cardiac monitoring was recommended due to the progressive nature of the syndrome.
Prominent vertebrae, winging of the scapula, and a loose skin fold
X-ray pelvis shows congenital hip dysplasia
X-ray spine shows increased intervertebral disc spaces
Discussion
The management of Marfan syndrome is complex and necessitates a multidisciplinary approach to address the diverse clinical manifestations affecting multiple organ systems. The primary goals of management involve preventing cardiovascular complications, optimizing musculoskeletal health, and addressing ocular abnormalities. A tailored and comprehensive care plan involving a team of specialists is essential to improve the quality of life for individuals with Marfan syndrome [7]. Cardiovascular management includes regular cardiac monitoring in individuals with Marfan syndrome due to the inherent risk of aortic root dilation and aortic dissection. Beta-blockers, such as propranolol or atenolol, are commonly prescribed to reduce the rate of aortic dilation and lower the risk of dissection. Angiotensin receptor blockers may also be considered, either alone or in combination with beta-blockers, for those who cannot tolerate beta-blockers or require additional blood pressure control. Surgical intervention, such as aortic root replacement, may be indicated in severe cases to prevent catastrophic cardiovascular events [8]. Musculoskeletal management involves managing orthopedic complications, including scoliosis, joint hypermobility, and pectus deformities, which are common in Marfan syndrome. Physical therapy and regular exercise are crucial in improving musculoskeletal function and preventing complications. Orthopedic interventions, such as bracing for scoliosis or surgical correction of pectus deformities, may be considered based on the severity of symptoms and functional impairment. Regular orthopedic follow-up is essential to monitor skeletal development and intervene as needed [9]. Ocular management involves ophthalmologic evaluation in individuals with Marfan syndrome to detect and manage ocular manifestations such as lens dislocation, myopia, and retinal detachment. Corrective lenses and surgery may be recommended to address visual impairment. Routine eye examinations are essential for ongoing monitoring and early intervention [10]. Genetic counseling is integral for individuals with Marfan syndrome and their families to understand the genetic basis of the condition, discuss inheritance patterns, and make informed family planning decisions. Psychosocial support is also crucial, as living with a chronic condition can impact mental well-being. Support groups and counseling services can provide valuable resources for individuals and their families [8-10].
Conclusions
The management of Marfan syndrome demands a comprehensive and coordinated approach to address its diverse clinical manifestations. A multidisciplinary team involving cardiologists, orthopedic specialists, ophthalmologists, and genetic counselors is essential for effectively caring for individuals with Marfan syndrome. Cardiovascular complications can be mitigated by emphasizing regular cardiac monitoring, medical interventions, and surgical procedures when necessary. Musculoskeletal management, encompassing physical therapy, exercise, and orthopedic interventions, aims to optimize functional outcomes. Ocular assessments and interventions contribute to maintaining visual health. Furthermore, genetic counseling and psychosocial support play pivotal roles in providing information, facilitating informed decision-making, and addressing the emotional aspects of living with a chronic genetic condition. By combining medical expertise, ongoing surveillance, and individualized care plans, the management of Marfan syndrome strives to enhance the quantity and quality of life for affected individuals. Continued research and awareness efforts are crucial for refining therapeutic strategies and ensuring the best possible outcomes for those living with this complex genetic disorder.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Marfan syndrome Radiol Technol Chaffins JA 222236782007 https://pubmed.ncbi.nlm.nih.gov/17242442/17242442 · pubmed ↗
- 2Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene Nature Dietz HC Cutting GR Pyeritz RE 3373393521991185220810.1038/352337 a 0 · doi ↗ · pubmed ↗
- 3The importance of mutation detection in Marfan syndrome and Marfan-related disorders: report of 193 FBN 1 mutations Hum Mutat Comeglio P Johnson P Arno G 92828200710.1002/humu.950517657824 · doi ↗ · pubmed ↗
- 4Marfan's syndrome Lancet Judge DP Dietz HC 1965197636620051632570010.1016/S 0140-6736(05)67789-6PMC 1513064 · doi ↗ · pubmed ↗
- 5Marfan syndrome revisited: from genetics to the clinic Rev Port Cardiol (Engl Ed) Coelho SG Almeida AG 2152263920203243910710.1016/j.repc.2019.09.008 · doi ↗ · pubmed ↗
- 6Valvular disease in marfan syndrome: surgical considerations and management Curr Cardiol Rep Plichta RP Glower DD Hughes GC 232120193082874910.1007/s 11886-019-1110-3 · doi ↗ · pubmed ↗
- 7Cardiovascular manifestations in Marfan syndrome and related diseases; multiple genes causing similar phenotypes Clin Genet Cook JR Carta L Galatioto J Ramirez F 11208720152486716310.1111/cge.12436 · doi ↗ · pubmed ↗
- 8The musculoskeletal manifestations of Marfan syndrome: diagnosis, impact, and management Curr Rheumatol Rep Pollock L Ridout A Teh J 812320213482599910.1007/s 11926-021-01045-3PMC 8626407 · doi ↗ · pubmed ↗