Insights from the SNP analysis of TYMP gene linking MNGIE
Najat Sifeddine, Lamiae Elkhattabi, Chaimaa Ait El Cadi, Al Mehdi Krami, Khadija Mounaji, Bouchra el khalfi, Abdelhamid Barakat

TL;DR
This study analyzes TYMP gene mutations linked to MNGIE syndrome, identifying harmful SNPs that affect protein structure and function.
Contribution
The study introduces a novel approach combining predictive algorithms and 3D modeling to identify harmful TYMP gene variants.
Findings
119 potentially deleterious nsSNPs were identified, with 82 in highly conserved regions.
79 nsSNPs were found to reduce TP protein stability.
3D analysis of 18 nsSNPs revealed altered amino acid interactions affecting protein function.
Abstract
TYMP gene, which codes for thymidine phosphorylase (TP) is also known as platelet-derived endothelial cell growth factor (PD-ECGF). TP plays crucial roles in nucleotide metabolism and angiogenesis. Mutations in the TYMP gene can lead to Mitochondrial Neurogastrointestinal Encephalopathy (MNGIE) syndrome, a rare genetic disorder. Our main objective was to evaluate the impact of detrimental non-synonymous single nucleotide polymorphisms (nsSNPs) on TP protein structure and predict harmful variants in untranslated regions (UTR). We employed a combination of predictive algorithms to identify nsSNPs with potential deleterious effects, followed by molecular modeling analysis to understand their effects on protein structure and function. Using 13 algorithms, we identified 119 potentially deleterious nsSNPs, with 82 located in highly conserved regions. Of these, 53 nsSNPs were functional and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
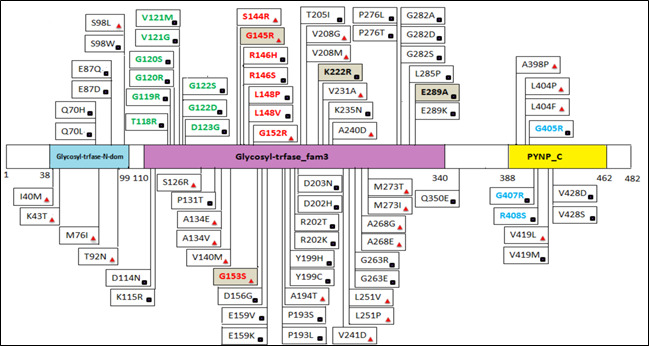 Figure 1
Figure 1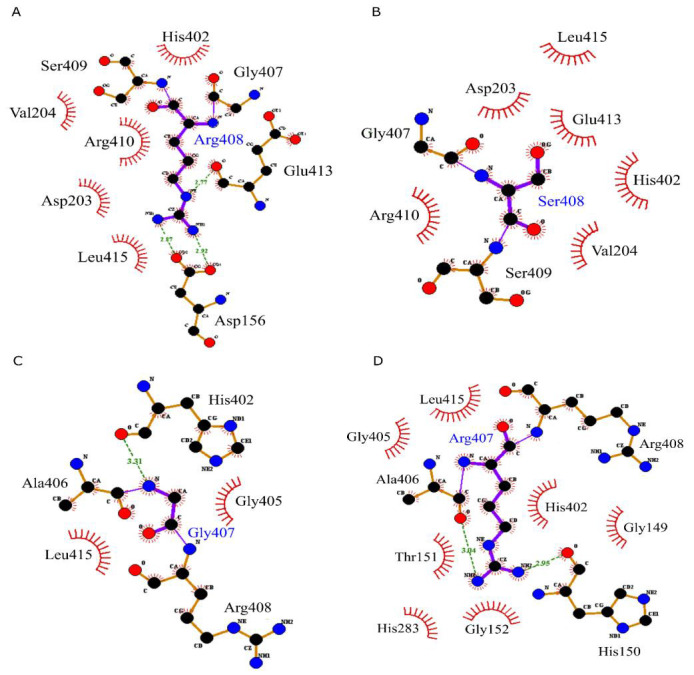 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMitochondrial Function and Pathology · Metalloenzymes and iron-sulfur proteins · Cancer, Hypoxia, and Metabolism
Background:
The TYMP gene, responsible for producing thymidine phosphorylase (TP), is situated on chromosome 22q13.33 [1]. The TP, also known as platelet-derived endothelial cell growth factor, is an enzyme that plays a crucial role in catalyzing the reversible phosphorolysis of thymidine, deoxyuridine, and their analogs (excluding deoxycytidine). This enzymatic activity leads to the formation of the corresponding bases and 2-deoxy-D-ribose-1-phosphate (2-dR-1-P) [2]. In the human body, the expression of TP, also known as hTP, is noteworthy in several tissues, including macrophage-like cells, the placenta, lymph nodes, spleen, liver, lungs, and peripheral lymphocytes [3]. The TYMP is found to be overexpressed in various cancer types, encompassing head and neck [4], breast [5], lung [6], oral squamous carcinoma [7], esophageal [8], gastric [9], colorectal [10], bladder [11], prostate [12], ovarian [13], and cervical [14] cancers, among several others. Its biological effects in cancer are primarily characterized by strong pro-angiogenic [15] properties and anti-apoptotic activity [16]. Mutations within the TP gene are an uncommon source of mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) [17]. Patients diagnosed with MNGIE display a marked reduction in TP activity, accompanied by a pronounced increase in the levels of thymidine and deoxyuridine in both the blood and tissues. This elevated presence of these substances has detrimental effects, causing disruption of mitochondrial DNA [1]. MNGIE presents a clinical profile characterized by a spectrum of symptoms, encompassing ptosis, ophthalmoparesis, gastrointestinal dysmotility, cachexia, peripheral neuropathy, myopathy, leukoencephalopathy, and lactic acidosis. Typically, the onset of MNGIE disease manifests before the age of 30 and sadly leads to the premature mortality of affected individuals between the ages of 20 and 40 [18]. This condition is intricately associated with the depletion and deletion of mitochondrial DNA (mtDNA), resulting from abnormalities in mitochondrial nucleoside/nucleotide metabolism [19]. Nonsynonymous SNPs (nsSNPs) located in coding regions can induce alterations in protein structure and/or function. Furthermore, in untranslated regions (UTRs), they frequently correlate with a range of diseases [20]. Identification of deleterious nsSNPs for most Human genes remains a major challenge in medical genetics. Therefore, it is of interest to identify deleterious SNPs that may affect the TP protein structure and/or function. In silico analyses conducted in this study not only advance our understanding of the impact of deleterious SNPs on TP protein structure and function but also lay a solid foundation for future experimental validations.
Materials and Methods:
Collection of nsSNPs:
Information regarding single nucleotide polymorphisms (SNPs) within the human TP gene was sourced from Ensembl (ensembl.org/), while the FASTA amino acid sequence of the TP protein (P19971) was retrieved from the UniProt database [21].
Prediction of protein alterations:
The pathogenicity of each non-synonymous SNP (nsSNP) collected was predicted using PredictSNP [22], a resource consolidating predictions from various tools including SIFT (Sorting Intolerant from Tolerant) [23] , PolyPhen-2 (Polymorphism Phenotyping v2) [24], PhD-SNP [25], PANTHER [26], and SNAP [27]. SIFT employs sequence homology to predict the impact of coding mutations on protein function, while PolyPhen-2 assesses the influence of substitutions on protein structure and function based on physical properties. PhD-SNP utilizes support vector machine (SVM) methods to classify mutations as disease-causing or benign. PANTHER predicts pathogenicity based on evolutionary patterns. MAPP [24] predictions were based on physicochemical variation in sequence alignments.
Sequence conservation:
ConSurf [28], a web-based algorithm, was employed to predict functionally important regions of the protein by estimating the degree of conservation of amino acid sites based on homology. The given score is between 1 and 9, representing the level of conservation of each amino acid. A score of 9 represents a highly conserved region, a score of 1 represents a highly variable region, and a score of 5 represents the average. This tool also reveals the type of residue in the giving position of the protein, which can be functional or structural and buried or exposed.
Prediction of nsSNPs positions in different protein domains:
The InterPro tool [29] facilitates the prediction of domains and important sites of proteins based on functional analysis and classification into families. In this study, the InterPro tool was utilized to identify the positions of nsSNPs within different protein domains.
System preparation and structural analysis:
The X-ray crystal structure of the human TP protein bound with thymine was retrieved from the Protein Data Bank (PDB) with a resolution of 2.31 Å (PDB ID 2j0f) [30]. Mutant protein structures were generated by substituting amino acids at corresponding positions, followed by energy minimization using the SPDB viewer tool [31] based on the GROMOS 96 force field.
Prediction of the effect of nsSNPs located in the UTR region:
The 5' and 3' untranslated regions (UTRs) play crucial roles in post-transcriptional gene regulation, translation efficiency, mRNA subcellular localization, and stability. UTRScan [32] was employed to predict functional SNPs within these regions. This tool searches submitted sequences for motifs present in UTRsite, which derives data from UTRdb, a curated database updated through primary data mining and experimental validation.
Results:
SNP datasets:
A total of 513 non-synonymous SNPs (nsSNPs) were retrieved from the thymidine phosphorylase (TP) gene data available in Ensembl. Among these, 124 SNPs were identified in the 5' untranslated region (UTR), and 23 were located in the 3' UTR of the human TP gene.
Prediction of deleterious nsSNPs:
Out of 513 nsSNPs, 119 were predicted as deleterious by all integrated tools in PredictSNP and was selected for further analysis (Table 1).Conservation analysis using the Consurf web server revealed that out of 119 nsSNPs analyzed, 82 were located in highly conserved positions. Of these, 53 were identified as functional and exposed residues, while 29 were predicted to be buried. We selected only residues with a high degree of conservation (Figure 1).
Prediction of different domains in TP:
The InterPro tool identified three domains within the TP protein: Glycosyl-Transferase-N-Domain (38-99), Glycosyl-Transferase-Fam3 (110-340), and PYNP-C (388-462). The distribution of highly conserved nsSNPs within these domains is illustrated in Figure 1.
Impact of predicted deleterious mutations on tp stability:
Using I-Mutant 20, DUET, and MUpro web servers, it was found that 79 nsSNPs led to a decrease in the stability of TP. Table 2 summarizes these results.
Structural analysis:
18 deleterious nsSNPs were selected for investigation. These chosen nsSNPs encompassed three variants located within residues crucial for thymine binding (R202K, R202T, and T118R), eight situated proximally to the active site (G120R, G120S, V121G, G122D, G122S, D123G, V208G, and V241D), two positioned within the loop involved in the closed conformation and stabilization of the dimer interface (G407R and R408S), and eight nsSNPs identified within the phosphate-binding site (S144R, G145R, R146H, R146S, and G153S). The substitution of arginine with threonine at position T118 resulted in the formation of a covalent bond with the thymine ligand and alterations in hydrophobic and hydrogen interactions compared to the native TP form. Mutations R202K and R202T led to the loss of hydrogen bonds with the thymine ligand and significant variations in hydrophobic interactions compared to the native form. The replacement of valine with guanine at conserved position 208 disrupted the hydrophobic interaction network compared to native TP. The V241D variant displayed destabilization in the hydrophobic domains, characterized by the acquisition of hydrophobic bonds with thymine and the loss of an interaction with IL241 compared to native TP. Additionally, the eight nsSNPs (S144R, G145R, R146H, R146S, L148P, L148V, G152R, and G153S) located in the phosphate-binding site, a highly conserved region, induced changes in both hydrophobic and hydrogen interactions.
Prediction of deleterious nsSNPs in UTRs:
Using the UTRscan server, 32 SNPs were predicted to be functional in the internal ribosome entry site (IRES) within the 5' UTR of the TP gene. These functional SNPs are listed in Table 3.
Discussion:
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is an uncommon autosomal recessive disorder that arises from mutations in the TP gene, causing dysfunction of the TP enzyme. TP functions as a homodimeric enzyme, with each subunit consisting of an α-helical domain (α domain) and a substantial α/β domain. These two domains are separated by a significant cleft that accommodates the active site for substrate binding [33]. Multiple reported missense mutations in the TP gene have been associated with the development of MNGIE [34]. A non-synonymous single nucleotide polymorphism (nsSNP) refers to a single-base alteration within the coding region of a gene, leading to the substitution of one amino acid for another in the corresponding protein. Investigating nsSNPs with functional relevance to diseases is a crucial goal in the fields of human molecular biology and medical research. Nevertheless, the sheer abundance of identified SNPs poses challenges in elucidating their biological significance through traditional wet laboratory experiments [35]. Over the past few decades, many studies have employed computational methods to assess the influence of mutations on protein structure and function. These approaches are effective in predicting whether a single-nucleotide polymorphism (SNP) has the potential to lead to a disease. In this work, different computational tools were used to identify the impact of nsSNPs on TP structure and stability. The total of 513 nsSNPs was analyzed by PredictSNP, and 119 of them were predicted to be the most deleterious. Additionally, we found 82 nsSNPs in highly conserved positions, including 54 nsSNPs in functional residues and 28 nsSNPs in structural residues.
The examination of the relationship between predicted amino acid alterations and the thermodynamic stability of proteins, as well as their impact on cellular stability and pathogenicity, implies that a decrease in stability may play a crucial role in the onset and progression of inherited diseases. It has been suggested that variants leading to the destabilization of proteins can disrupt their normal cellular functions, potentially giving rise to various genetic disorders [36]. In this study, the stability analysis revealed that the 79 nsSNPs reduced protein stability according to all the prediction tools employed. Only 18 deleterious nsSNPs were selected for molecular analysis based on their localization in the TP domains. This analysis focuses on the comparison of the differences in hydrogen bonds and hydrophobic interactions between the amino acids of the wild-type protein and its mutated forms. The deleterious SNPs (T118R, R202K, and R202T) contribute to thymine binding (Figure 2) and reveal a disruption of thymine binding (Table 3). The T118R mutation does not form a covalent bond with thymine. Instead, this mutation is associated with TP dysfunction, which can lead to the accumulation of thymine and other metabolites. This accumulation, caused by impaired TP enzymatic function, can contribute to the onset of MNGIE disease.
Deleterious nsSNPs localized in close proximity to the binding site of TP (G120R, G120S, V121G, G122D, G122S, D123G, V208M, V208G, and V241D) are in highly conserved positions. decrease TP's stability, leading to the disruption of the hydrogen and hydrophobic interaction, which may induce a change in the conformation of TP and may affect protein function. The two highly conserved nsSNPs (G407R and R408S) were localized in the important loop, which could potentially contribute to the integrity and stability of the closed conformation [37]. The structural property comparison between mutant forms and the WT protein showed a large change in the hydrophobic and hydrogen interactions (Figure 2). The residues Arg408, Ser409, and Arg410 make hydrogen bonds between the loop and the rest of the protein. Consequently, the two deleterious mutations are most likely to disrupt the structural and functional features of the WT protein. Eight nsSNPs (S144R, G145R, R146H, R146S, L148P, L148V, G152R, and G153S) are located in the glycine-rich loop, which has an important role in the binding of the catalytic phosphate [38]. Our in silico tests showed that the eight highlighted mutations are in a highly conserved region, decrease the TP's stability, and cause a vast variation in the residue-neighbor interaction compared to the native form (Table 2). We suggest that the studied mutations overall could affect TP catalytic efficiency through two possible mechanisms: decreasing the structural stability of the protein and reducing its binding affinity towards the essential cofactor PI. In addition, phosphate-binding domains of TP are responsible for the initiation of the closed conformation of the active site [39]. We suggested that substitution of glycine with either arginine or serine may cause MNGIE disease occurrence by disrupting phosphate binding or rendering the TP catalysis less effective. Multiple experimental studies involving the TYMP gene have demonstrated that some of the 19 non-synonymous single nucleotide polymorphisms (nsSNPs) are associated with significant manifestations in subjects of diverse ages and phenotypes. For instance, the R202T mutation was discovered in the TP gene of a 55-year-old Dutch woman who presented with ophthalmoplegia, severe bilateral ptosis, muscle atrophy (while maintaining normal muscle strength), intact sensory testing, and hypoactive or absent tendon reflexes. Additionally, she exhibited extensive leukoencephalopathy and polyphasic potentials in her leg muscles [40]. Similarly, the V208M mutation has been described in a 61-year-old Anglo-American woman who presented with a complex array of health issues, including pancreatitis, small intestine ileus, recurrent nausea with vomiting, early satiety, borborygmi, colonic diverticulosis, and hepatopathy. Furthermore, she experiences demyelinating sensorimotor polyneuropathy and has developed ptosis, progressive external ophthalmoplegia (PEO), optic atrophy, hearing loss, patchy leukoencephalopathy, short-term memory disturbances, occasional inappropriate behaviors, insulin-dependent diabetes mellitus, and renal cell carcinoma identified in patients who met the clinical criteria for mitochondrial neurogastrointestinal encephalomyopathy [39]. Moreover, the G145R mutation has been identified in patients presenting clinical symptoms consistent with mitochondrial neurogastrointestinal encephalomyopathy, originating from different regions, including Israel and Puerto Rico. Similarly, the G153S mutation has been identified in affected patients with clinical symptoms consistent with mitochondrial disorders [19]. All of the experimental data strongly align with the findings from our bioinformatic study, thus providing comprehensive evidence for and an explanation of the impact of deleterious nsSNPs on the TYMP gene. The IRES signal was described as a distinct RNA region that directly promotes the binding of 40S ribosomal subunits to mRNA without previous scanning [40]. The impairment of IRES sequences can deregulate mRNA translation and lead to various diseases or disease susceptibilities. Such as Charcot-Marie-Tooth disease (CMTX) [41], multiple myeloma [42], and Fragile X syndrome (FXS) [43]. We defined 32 SNPs in the IRES region; these SNPs may impair TP synthesis and lead to disease. The early detection of these potentially deleterious mutations in the TP gene could enable preventive intervention for individuals at risk, thereby paving the way for a reduction in the prevalence of MNGIE.
Conclusion:
We identified 119 deleterious nsSNPs within the coding region of the TP gene. Out of them, 79 nsSNPs were predicted to perturb protein stability. Moreover, the structural analysis of 18 SNPs revealed disruption of the network of interaction compared to the native form of TP, which could destabilize the TP-Thymine complex and consequently induce the occurrence of the MNGIE disease. Additionally, we identified 32 functional SNPs in the 5' UTR, which could affect protein synthesis and may lead to diseases. This study lays the groundwork for future research aimed at experimentally validating the predictions of our in silico analysis, thereby paving the way for a better understanding of the underlying mechanisms of MNGIE.
Data availability
All the datasets and structures generated for this study are available from the authors.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Nishino I Science 1999283689992402910.1126/science.283.5402.689 · doi ↗ · pubmed ↗
- 2Friedkin M Roberts DJ Biol Chem. 195420724513152099 · pubmed ↗
- 3Yoshimura A Biochim Biophys Acta 19901034107232825510.1016/0304-4165(90)90160-x · doi ↗ · pubmed ↗
- 4Giatromanolaki A Clin Exp Metastasis 199816665993261310.1023/a:1006554512338 · doi ↗ · pubmed ↗
- 5Ruckhäberle E Eur J Cancer 2010465492002248610.1016/j.ejca.2009.11.020 · doi ↗ · pubmed ↗
- 6Giatromanolaki AJ Pathol 1997181196912072510.1002/(SICI)1096-9896(199702)181:2<196::AID-PATH 763>3.0.CO;2-U · doi ↗ · pubmed ↗
- 7Ranieri G Int J Oncol 200221131712429983 · pubmed ↗
- 8Lee S Br J Cancer 20101038452070012510.1038/sj.bjc.6605831 PMC 2966625 · doi ↗ · pubmed ↗