Carbon–nitrogen transmutation in polycyclic arenol skeletons to access N-heteroarenes
Hong Lu, Yu Zhang, Xiu-Hong Wang, Ran Zhang, Peng-Fei Xu, Hao Wei

TL;DR
Scientists developed a new method to convert polycyclic arenols into N-heteroarenes by swapping carbon atoms with nitrogen, enabling the creation of complex aromatic structures.
Contribution
A novel skeletal editing strategy for carbon-to-nitrogen transmutation in polycyclic arenol frameworks is introduced.
Findings
The method enables selective nitrogen insertion into C–C bonds of arenol frameworks via azidative dearomatization and aryl migration.
The approach allows streamlined assembly of complex polycyclic heteroaromatics with broad functional group tolerance.
Transformations of the products showed potential in materials chemistry, including synthesis of complex biheteroarene skeletons.
Abstract
Developing skeletal editing tools is not a trivial task, and realizing the corresponding single-atom transmutation in a ring system without altering the ring size is even more challenging. Here, we introduce a skeletal editing strategy that enables polycyclic arenols, a highly prevalent motif in bioactive molecules, to be readily converted into N-heteroarenes through carbon–nitrogen transmutation. The reaction features selective nitrogen insertion into the C–C bond of the arenol frameworks by azidative dearomatization and aryl migration, followed by ring-opening, and ring-closing (ANRORC) to achieve carbon-to-nitrogen transmutation in the aromatic framework of the arenol. Using widely available arenols as N-heteroarene precursors, this alternative approach allows the streamlined assembly of complex polycyclic heteroaromatics with broad functional group tolerance. Finally, pertinent…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
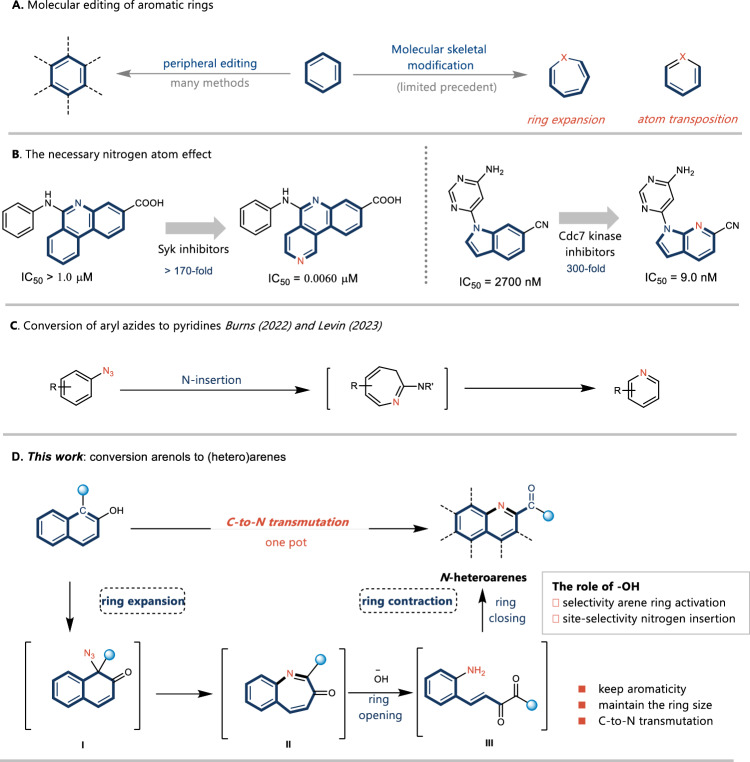 Figure 1
Figure 1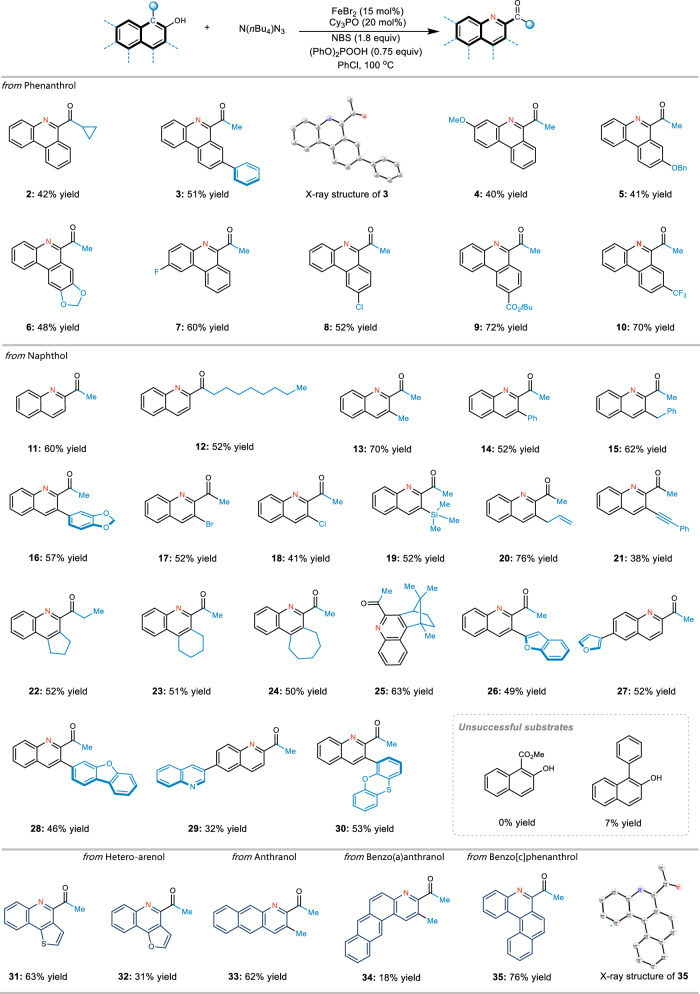 Figure 2
Figure 2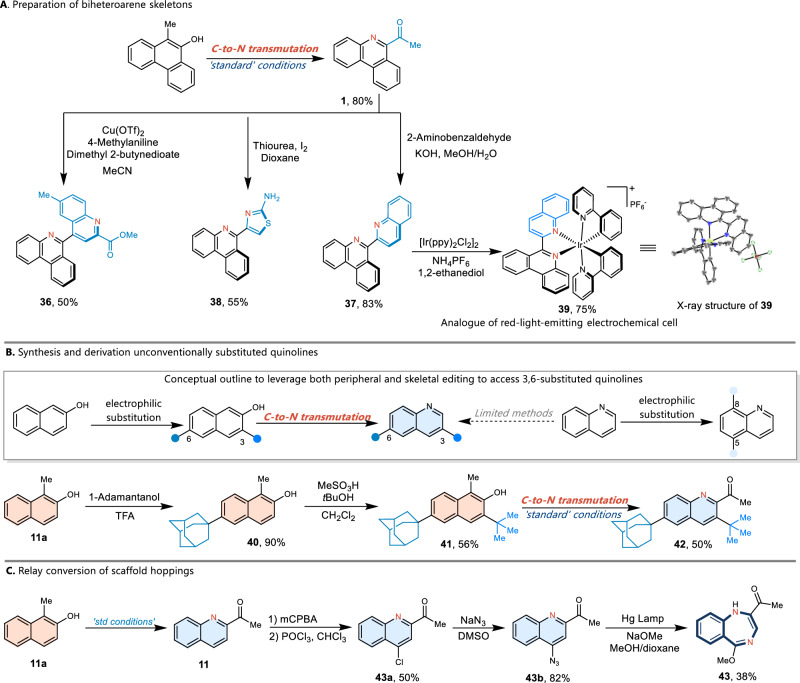 Figure 3
Figure 3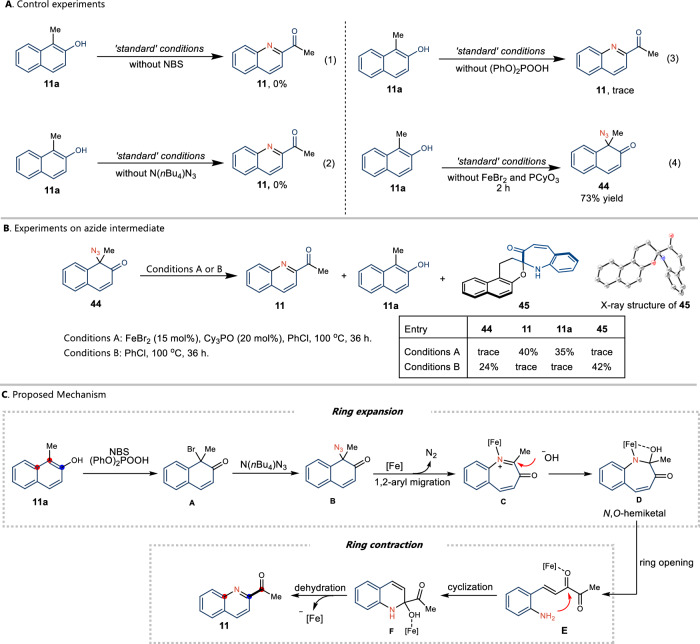 Figure 4
Figure 4- —https://doi.org/10.13039/501100001809National Natural Science Foundation of China (National Science Foundation of China)
- —Natural Science Basic Research Plan for Distinguished Young Scholars in Shaanxi Province of China (2022JC-08)
- —Natural Science Basic Research Plan in Shaanxi Province of China (2023-JC-YB-126)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSpanish Philosophy and Literature · History of Education in Spain · Spanish Literature and Culture Studies
Introduction
Organic synthesis underpins the evolution and advancement of broad areas of science, from materials to medicine. Arenes are among the most widely used rings in medicine and natural products. The functionalization of arenes is a particularly attractive tool for the production of pharmaceuticals, natural products, and molecular materials^1–4^. However, their application has so far been largely focused on C–H functionalization chemistry (peripheral editing), and the precise modification of the aromatic ring skeleton remains largely unexplored (Fig. 1A)^5–7^. Single-atom skeletal editing has become an extremely powerful tool for straightforwardly modifying the core skeleton of organic molecules. Recently, a limited number of single–atom insertion or deletion reactions have been developed to reshape the underlying molecular skeletons^8–21^. However, the direct modification of valuable core structures by replacing one atom in a ring system without changing the ring size and aromaticity remains elusive^22–31^, although it has been recognized as a highly desirable transformation.Fig. 1. Examples of carbon–nitrogen transmutation and our reaction design.A Molecular editing of aromatic rings. B Examples of the necessary nitrogen atom effect. C Conversion of aryl azides to pyridines. D This study.
One of the most studied among N-heteroarenes are pyridines, which serve as a bioisosteric replacement of benzene counterparts within the parent molecules^32–34^. The replacement of carbon with nitrogen in aromatic ring systems can have several important effects on the molecular and physicochemical properties relevant to multiparameter optimization (Fig. 1B). This necessary nitrogen atom effect is a versatile high-impact design element for multiparameter optimization, which has been shown to improve various key pharmacological parameters^35^. Recently, Burns and Levin independently reported groundbreaking methods for the direct conversion of arenes to pyridines via nitrene internalization (Fig. 1C)^36,37^. In these process, additional steps for installation and isolation of aryl azides are always requried, which indicated that a selective, and straightforward transformation of diverse arenes into N-heteroarenes remains an important goal^38^.
A key challenge in this transformation is the stability of the aromatic compounds. Our design overcomes this intrinsic challenge using arenols as substrates. Dearomatization of arenols disrupts the stability of the aromatic ring and promotes subsequent skeletal transformations^39–48^. Arenol can also act as a selectivity controlling element in site-selective skeletal editing. Our group’s recent work employing this dearomative strategy to promote ring expansion of arenols inspired us to continue investigating this strategy to more complex skeletal editing transformation^49^. In this work, we describe the direct carbon-to-nitrogen transmutations in arenols. This reaction involves two stages: ring expansion and contraction (Fig. 1D). In the first stage, the insertion of nitrogen atoms is achieved by azidative dearomatization of an arenol and intramolecular aryl migration. In the second stage, a carbon atom moves out of the ring skeleton through ring-opening, and ring-closing (ANRORC), which ultimately furnishes desired carbon–nitrogen transmutation in polycyclic arenol skeletons^50–52^.
Results
Reaction optimization
We began our investigation using methylphenanthren-9-ol 1a as the reaction partner. (PhO)2_POOH, NBS, and N(nBu)4_N_3 were employed as reagents for the in situ formation of the azido ketone intermediate (see Supplementary Information, section 2.2.2). For optimization, we observed the formation of desired product 1 in 80% yield using FeBr_2 and Cy_3_PO as an effective catalyst–ligand combination in PhCl (Table 1, entry 1). A control experiment revealed that an iron salt was essential for obtaining the desired product (Table 1, entry 2). Other iron salts, including FeCl_2_, Fe(OTf)2, Fe(OAc)2, and Fe(acac)3, exhibited lower efficiency than inexpensive FeBr_2_ (Table 1, entries 3 − 6). Furthermore, when other established metal nitrenoid formation catalysts, including copper, rhodium, cobalt, and ruthenium, were used, the desired product was not obtained satisfyingly (Table 1, entries 7–10)^53,54^. Further optimization showed that this reaction was slightly improved using Cy_3_PO (Table 1, entry 11). The reaction appeared to be less sensitive to solvents, as replacing the PhCl with either toluene or THF furnished 1 in good yield (Table 1, entries 12 and 13). The yield decreased slightly when 10 mol% FeBr_2_ and 15 mol% Cy_3_PO were used (Table 1, entry 14).Table 1. Screening of reaction conditions.^a^EntryVariation from ‘standard conditions’Yield (%)^b^1None80 (61)^c^2w/o FeBr_2_trace3FeCl_2_ instead of FeBr_2_714Fe(OTf)2 instead of FeBr_2_755Fe(OAc)2 instead of FeBr_2_326Fe(acac)3 instead of FeBr_2_107Cu(OTf)2 instead of FeBr_2_<108[[CH_3_(CH_2_)6_CO_2]2_Rh]2 instead of FeBr_2_349TMOPP-Co instead of FeBr_2_trace10Ru_3(CO)13 instead of FeBr_2_<1011w/o Cy_3_PO7012In toluene7513In THF701410 mol% FeBr_2_ and 15 mol% Cy_3_PO instead61^a^Unless otherwise specified, all reactions were carried out using 1a (0.1 mmol), NBS (0.18 mmol), N(nBu_4_)N_3_ (0.3 mmol), (PhO)2_POOH (0.075 mmol), FeBr_2 (0.015 mmol), and Cy_3_PO (0.02 mmol) in PhCl (1.0 mL) at 100 °C for 36 h.^b^Isolated yields after chromatography.^c^Scale-up reaction by using 1.0 mmol of 1a.
Substrate scope
Considering the optimal reaction conditions, the substrate scope was determined (Fig. 2). Various arenols, including phenanthrol (1–10), naphthol (11–30), anthranol (33) benzo(a)anthranol (34), and benzo[c]phenanthrol (35), can effectively undergo the desired carbon–nitrogen transmutation. Both electron-rich and electron-deficient aromatic substrates were suitable for the process. It was found that the substituents at the ortho positions of the arenol are significant. When the substituent was an alkyl group, the corresponding arenols underwent atom transmutation smoothly in moderate-to-good yield and chemoselectivity. The presence of a phenyl group or an electron-withdrawing group such as CO_2_Me at the ortho-position can inhibit this reaction. However, various functional groups, such as ether (4 and 5), acetals (6 and 16), aryl halides (7, 8, 17 and 18), esters (9), trifluoromethyl (10), trimethylsilyl (TMS) (19), alkenes (20), and alkynes (21), were tolerated in this transformation. In addition, several naphthyl-fused rings (22–25) were suitable substrates, affording the desired products in moderate-to-good yields. Heterocyclic moieties such as benzofuran (26), furan (27), dibenzofuran (28), quinoline (29), and phenoxathiine (30) were also compatible. Moreover, fused heteroarenols such as naphtho[1,2-b]thiophene (31) and naphtho[1,2-b]furan (32) can be incorporated, providing pharmaceutically interesting fused-ring skeletons that are non-trivial to prepare. The structures of 3 and 35 were identified using X-ray crystallography.Fig. 2. Substrate scope of the carbon–nitrogen transmutation in polycyclic arenol.^a,b a^Isolated yields after chromatography are shown. ^b^ Reaction conditions: substrate (0.1 mmol), NBS (0.18 mmol), N(nBu_4_)N_3_ (0.3 mmol), (PhO)2_POOH (0.075 mmol), FeBr_2 (0.015 mmol), and Cy_3_PO (0.02 mmol) in PhCl (1.0 mL) at 100 °C for 36 h.
Synthetic utility
The successful development of the atom transmutation protocol offers a rapid and modular approach to access complex biheteroarene skeleton, a common structural motif found in bioactive compounds (Fig. 3A). Compound 37 could be easily transformed to the iridium complexes 39, which could serve as the red-light-emitting electrochemical cell^55,56^. Next, the synthetic versatility of the C-to-N transmutation was demonstrated through the preparation of 3,6-disubstituted quinolines 42, which could not be obtained from directly electrophilic substitution of quinolines (Fig. 3B). Specifically, the successful development of the carbon–nitrogen transmutation offers exciting opportunities to devise more complex skeletal editing transformations via combinations of atom insertions and deletions. Benzo[1,4]diazepine 43 can be accessed through a C–H azidation and aryl migration sequence from 11, presently the formal carbon deletion and two nitrogen insertion products of starting naphthol 11a (Fig. 3C).Fig. 3. Application potential of carbon–nitrogen transmutation.A Applications that allow access to complex biheteroarene skeletons. B Preparation of unconventionally 3,6-substituted quinolines. C Sequential skeletal editing transformations of naphthol.
Mechanistic considerations
To elucidate the mechanism of this transformation, series control experiments were first conducted. The reactions without addition of NBS or N(nBu_4_)N_3_ failed to produce the desired product (Fig. 4A, equations 1 and 2). And trace amount of 11 was observed when (PhO)_2_POOH was absent from the reaction mixture (Fig. 4A, equation 3). It’s worth noting that azide ketone 44 could be isolated in 73% yield in the absense of Fe catalyst after 2 h (Fig. 4A, equation 4). These results indicated that the proposed azidative dearomatization of arenol might be involved (Fig. 1D)^57^. The azide ketone 44 was then tested with and without the addition of the Fe catalyst (Fig. 4B). It was found that the desired product 11 was formed in 40% yield, and 35% yield of 11a was isolated in presence of Fe catalyst, which demonstrated that the proposed azidation is a reversible process via successive single-electron transfer (SET) from Fe(II) to eliminate azide^58^. On the contrary, only trace amount of product 11 and 11a was observed without Fe catalyst. And a byproduct 45 was detected in 42% yield and recovered 44 in 24%^59^. These results revealed that the Fe catalyst is not only involved in aryl migration, but is also essential for the ring contraction process^60^.Fig. 4. Mechanistic studies.A Control experiments. B Control experiments using azide intermediate showing that Fe catalyst is essential for aryl migration and ring contraction process. C Proposed mechanism.
Based on the literature reports and our observations, a plausible mechanism is proposed (Fig. 4C). Initially, the N-bromosuccinimide-mediated dearomatization of the corresponding naphthol of 11a afforded the brominated ketone intermediate A, which subsequently reacted with N(nBu)4_N_3 to generate azido ketone B. Then iron salt reacted with B can form metal−nitrene species, which would then undergo 1,2-aryl migration to form ring expansion intermediate C. Subsequently, addition of hydroxide anion to the imine group of C induces N,O-hemiketal D. The collapse of D with assistance of iron salt produces ring-opening amino-ketone intermediate E^61,62^, which undergoes re-cyclization and dehydration to form stable benzoquinoline 11 and release Fe catalyst.
In conclusion, this study proposed a unique strategy that enables straightforward carbon-to-nitrogen transmutations in arenols through a one-pot ring expansion-contraction sequence. This site-selective atom transformation is based on sequentially combining three transformations in one pot using aryl migration and imine transposition as key steps and opens new opportunities for single-atom skeletal edit design. Further preparation of complex biheteroarene skeleton and unconventionally substituted quinoline highlights the potential of this study. This provides an alternative for the development of N-heteroarenes and demonstrates significant potential in materials chemistry.
Methods
General condition for carbon–nitrogen transmutation
Substrate (0.1 mmol), NBS (0.12 mmol), N(nBu_4_)N_3_ (0.2 mmol), FeBr_2_ (0.015 mmol), tricyclohexylphosphine oxide (0.02 mmol), (PhO)2_POOH (0.05 mmol), and PhCl (1.0 mL) was successively added to an 10 mL sealed tube equipped with a Teflon-coated magnetic stir bar. The tube then was sealed with a Teflon screw cap and placed on a hotplate pre-heated to 100 °C with vigorous stirring. After 18 h, the reaction was cooled to room temperature and another portion of NBS (0.06 mmol, 0.6 equiv), N(nBu_4)N_3 _(0.1 mmol) and (PhO)_2_POOH (0.025 mmol) was successively added to the sealed tube. The tube then reacted at 100 °C with vigorous stirring. After 18 h, the reaction was cooled to room temperature. The solvent was evaporated and the residue was directly purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate = 20/1) to give the desired products.
Supplementary information
Supplementary Information Peer Review File
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Engle KM Mei T-S Wasa M Yu J-Q Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions Acc. Chem. Res.20124578880210.1021/ar 200185 g 22166158 PMC 3334399 · doi ↗ · pubmed ↗
- 2Neufeldt SR Sanford MS Controlling site selectivity in palladiumcatalyzed C–H bond functionalization Acc. Chem. Res.20124593694610.1021/ar 300014 f 22554114 PMC 3378812 · doi ↗ · pubmed ↗
- 3Wencel-Delord J Glorius FC–H bond activation enables the rapid construction and late-stage diversification of functional molecules Nat. Chem.2013536937510.1038/nchem.160723609086 · doi ↗ · pubmed ↗
- 4Abrams DJ Provencher PA Sorensen EJ Recent applications of C–H functionalization in complex natural product synthesis Chem. Soc. Rev.2018478925896710.1039/C 8CS 00716 K 30426998 · doi ↗ · pubmed ↗
- 5Jurczyk J Single-Atom Logic for Heterocycle Editing Nat. Synth.2022135236410.1038/s 44160-022-00052-135935106 PMC 9355079 · doi ↗ · pubmed ↗
- 6Liu, F., Anand, L. & Szostak, M. Diversification of indoles and pyrroles by molecular editing: New frontiers in heterocycle-to-heterocycle transmutation. Chem. Eur. J. 29, e 202300096 (2023).10.1002/chem.202300096 PMC 1019200636730110 · doi ↗ · pubmed ↗
- 7Zhaozhong L Paramasivam S Yongquan N Yong W Xihe B Skeletal editing of (hetero)arenes using carbenes Chem. Eur. J.202329 e 20230122710.1002/chem.20230122737230933 · doi ↗ · pubmed ↗
- 8Roque JB Kuroda YGöttemann LT Sarpong R Deconstructive diversification of cyclic amines Nature 201856424424810.1038/s 41586-018-0700-330382193 PMC 6317721 · doi ↗ · pubmed ↗