Comparative chloroplast genomes of Dactylicapnos species: insights into phylogenetic relationships
Shunquan Yang, Juntong Chen, Zhimin Li, Xianhan Huang, Xu Zhang, Qun Liu, Komiljon Tojibaev, Hang Sun, Tao Deng

TL;DR
This study analyzes the chloroplast genomes of seven Dactylicapnos species to understand their genetic structure and evolutionary relationships.
Contribution
The first comparative analysis of chloroplast genomes in Dactylicapnos, revealing structural and phylogenetic insights.
Findings
Chloroplast genomes of Dactylicapnos have a typical quadripartite structure with lengths between 172,344 bp and 176,370 bp.
Phylogenetic analysis grouped the seven species into three main clades corresponding to the three sections of the genus.
31 codons showed high synonymous codon usage in the chloroplast genomes of Dactylicapnos.
Abstract
Dactylicapnos is a climbing herbaceous vine, distributed from the Himalayas to southwestern China, and some of the species have important medicinal values. However, the chloroplast genomes of Dactylicapnos have never been investigated. In this study, chloroplast genomes of seven Dactylicapnos species covering all three sections and one informal group of Dactylicapnos were sequenced and assembled, and the detailed comparative analyses of the chloroplast genome structure were provided for the first time. The results showed that the chloroplast genomes of Dactylicapnos have a typical quadripartite structure with lengths from 172,344 bp to 176,370 bp, encoding a total of 133–140 genes, containing 88–94 protein-coding genes, 8 rRNAs and 37–39 tRNAs. 31 codons were identified as relative synonymous codon usage values greater than one in the chloroplast genome of Dactylicapnos genus based on…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
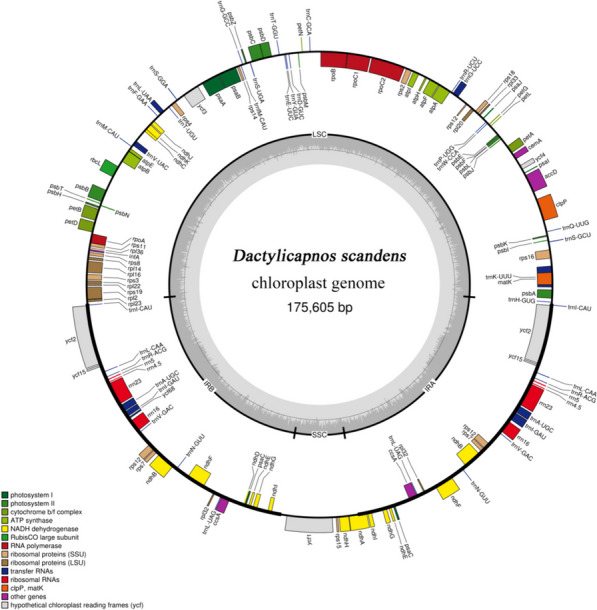 Figure 1
Figure 1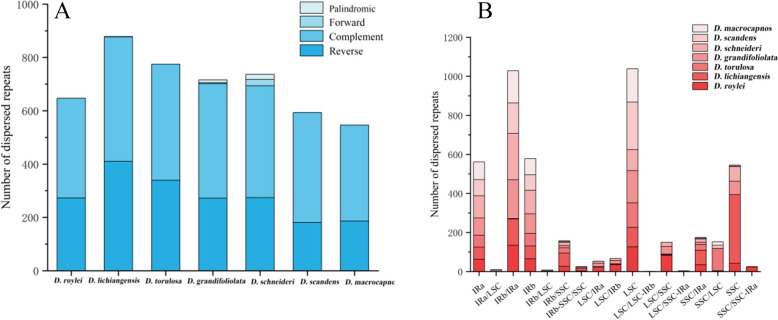 Figure 2
Figure 2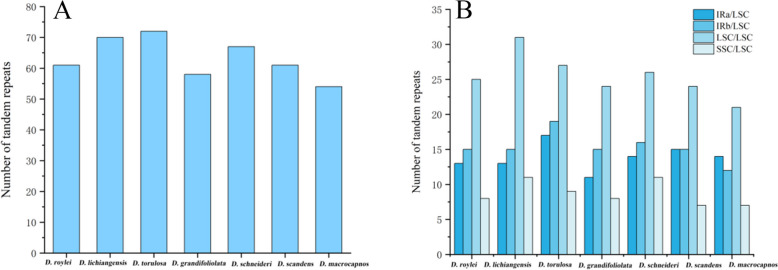 Figure 3
Figure 3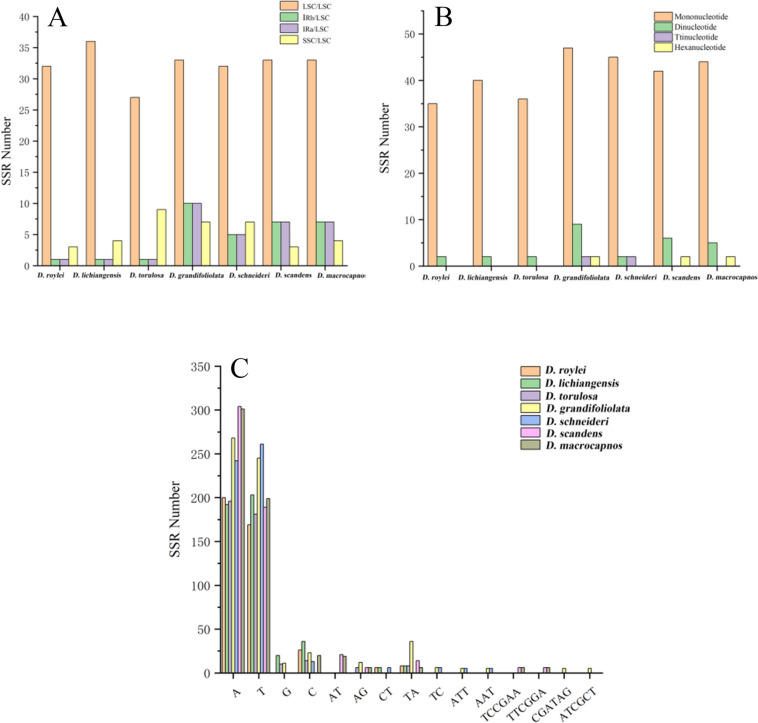 Figure 4
Figure 4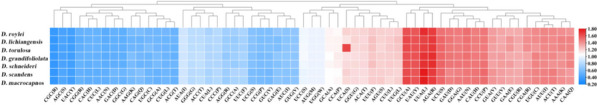 Figure 5
Figure 5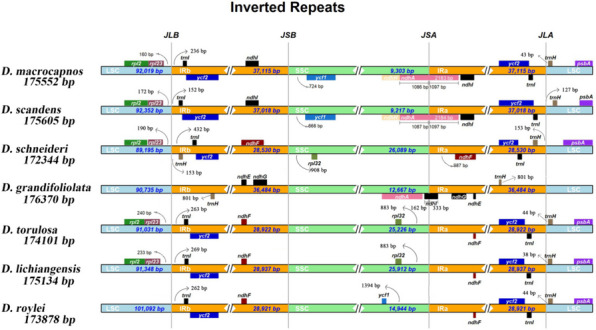 Figure 6
Figure 6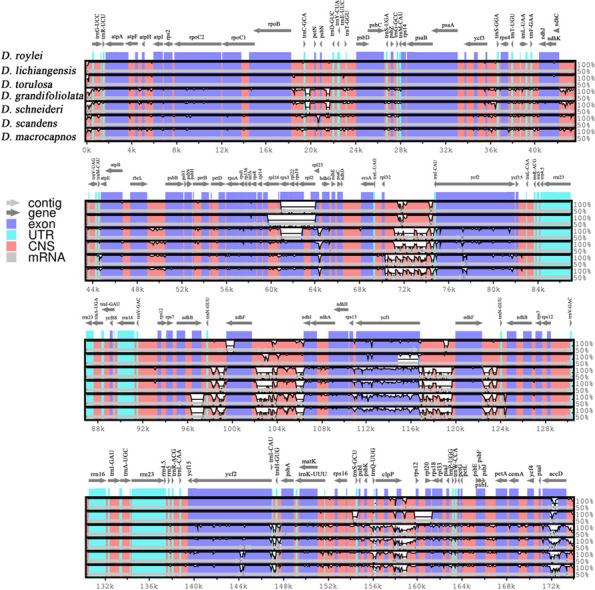 Figure 7
Figure 7 Figure 8
Figure 8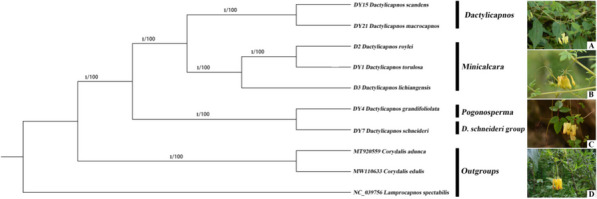 Figure 9
Figure 9- —National Natural Science Foundation of China
- —Second Tibetan Plateau Scientific Expedition and Research (STEP) program
- —Key R&D Program of Yunnan
- —Strategic Biological Resources Capacity Building Project of Chinese Academy of Sciences
- —Major Program for Basic Research Project of Yunnan Province
- —Key Projects of the Joint Fund of the National Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCephalopods and Marine Biology · Genetic diversity and population structure · Genomics and Phylogenetic Studies
Introduction
Dactylicapnos Wall. belongs to Fumarioideae (DC.) Endlicher. in the family Papaveraceae Juss., established by Wallich in 1826 [1]. There are about 15 species in the genus, distributed from the Himalayas to southwestern China [2–5]. Dactylicapnos is a climbing herbaceous vine, distinguished by its branched tendrils at the end of leaves, pendent raceme inflorescences, yellow bisymmetric flowers and subquadrangular stigma with a papilla on each corner [2]. Taxa of Dactylicapnos are rich in active ingredients such as isoquinoline alkaloids [6], with the highest content of isocorydine and protopine [7], and is used in a Bai Nationality folk medicine due to its analgestic, anti-inflammatory, hemostatic and anti-hypertensive effects [8].
Despite some species of Dactylicapnos are very important medicinal plants, the relationship between these species is not clear. Recent classification of Dactylicapnos by Lidén and Pathak [5] based on morphology divided the genus into three sections (sect. Dactylicapnos, sect. Minicalcara and sect. Pogonosperma) and two informal groups. Validity of these morphologically defined sections and informal groups could not be confirmed due to lack of a systematic molecular study of the genus. A few molecular studies performed to date included only a few species of the genus, Lidén [9] used rps16 fragments to explore the systematic relationship between Dicentra and Dactylicapnos, Pérez-Gutiérrez [10, 11] conducted a molecular phylogenetic study of the Fumarioideae using five plastid markers, which included only four Dactylicapnos species, and only three species were included in the study by Chen [12]. There has never been a systematic molecular study to resolve the genus infrageneric phylogeny based on cp genome. Thus, the monophyly of these morphologically defined sections and informal groups could not be confirmed to verify the Lidén and Pathak [5] classification.
Chloroplasts are important organelles for photosynthesis in green plants which genome uniparently inherited. The chloroplast (cp) genome size of angiosperms is in the range of 120 to 160 kb [13], with a typical quadripartite structure, consisting of two-copy inverted repeat (IR) of 20–28 kb, a large single-copy regon (LSC) of about 80–90 kb and a small single-copy region of 16–27 kb [14], usually encoding for 120–150 genes. It is known that the cp genome encodes all the tRNA and rRNA molecules and partial proteins required for its own function [15–17]. Due to the highly conservative structure, rich in genetic information [18], slow nucleotide substitution rate [19], and uniparental inheritance [20], the cp genome has been widely used in phylogenetics analyses and identifications [21]. At present, chloroplast genome sequences of many species have been published, but the species of Dactylicapnos has not been published yet. The lack of systematic molecular studies hampers the development and application of Dactylicapnos. This motivated the current study of a comparative genomic analysis of seven of Dactylicapnos species covering all three sections and one informal group, in order to understanding the evolution of the genus structure and clarification of the phylogenetic relationship in Dactylicapnos species.
Results
Chloroplast genome structure and characteristics analyses of Dactylicapnos species
The lengths of the studied cp genomes varied from 172,344 bp (Dactylicapnos schneideri (Fedde) Lidén.) to 176,370 bp (Dactylicapnos grandifoliolata Merrill.), with a typical quadripartite structure, a pair of IR regions (28,530 bp–37,115 bp), LSC regions (89,195 bp–101,092 bp) and SSC (9303 bp–26,089 bp) (Fig. 1; Table 1). In the studies species there was an identical level of GC content with the total content 40.0%–40.6%, 41.6%–43.6% in IR, 39.1%–39.4% in LSC, and 35.3%–38.0% in SSC. The GC content of IR region was higher than LSC and SSC.Fig. 1. Gene map of the chloroplast genomes of D. scandens. Genes inside the circle are transcribed clockwise, and those on the outside are transcribed counter-clockwise. Genes belonging to different functional groups have been color-coded. The darker grey area in the inner circle corresponds to GC content, while the lighter grey corresponds to AT contentTable 1The basic chloroplast genome information of seven Dactylicapnos speciesCharacteristicsD. macrocapnos**D. scandens**D. schneideri**D. grandifoliolata**D. torulosaD. lichiangensisD. royleiTotal length(bp)175,552175,605172,344176,370174,101175,134173,878LSC lenghth(bp)92,01992,35289,19590,73591,03191,348101,092IR lenghth(bp)37,11537,01828,53036,48428,92228,93728,921SSC lenghth(bp)9303921726,08912,66725,22625,91214,944Total numble of genes140140139139133133133Protein-coding genes94949292888888tRNA genes38383939373737rRAN genes8888888Overall GC content(%)40.0%40.0%40.2%40.2%40.6%40.6%40.6%GC content in LSC(%)39.1%39.1%39.1%39.1%39.4%39.4%39.3%GC content in IR(%)41.6%41.7%43.5%42.0%43.6%43.6%43.6%GC content in SSC(%)35.3%35.4%36.9%36.5%38.0%38.0%37.3%
The seven cp genomes have 133–140 genes, including 88–94 protein-coding genes, 37–39 tRNA genes, and 8 rRNA genes (Table 2). In the studied species, there are 18–26 genes with two copies, which were mostly comprised of seven protein-coding genes (ycf2, ycf15, ycf68, rps12, rps7, ndhB, ndhF), seven tRNA genes (trnI-CAU, trnL-CAA, trnR-AGC, trnA-UGC, trnI-GAU, trnV-GAC, trnN-GUU), and four rRNA (rrn5, rrn4.5, rrn23, rrn16), but the D. grandifoliolata also has six protein-coding genes (rpl32, ccsA, ndhD, psaC, ndhE, ndhG), two tRNA genes (trnH-GUG, trnL-UAG), D. schneideri also has one tRNA gene (trnH-GUG), and Dactylicapnos scandens Hutch. also have seven protein-coding genes (rpl32, ccsA, ndhD, psaC, ndhE, ndhG, ndhI) and one tRNA gene (trnL-UAG). Sixteen genes (trnG-UCC, atpF, rpoC1, trnL-UAA, trnV-UAC, petB, petD, rpl16, rpl2, trnA-UGC, trnI-GAU, rps12, ndhB, ndhA, trnK-UUU, rps16) contain a single intron, two genes (ycf3, clpP) have two introns, and the gene trnK-UUU has the largest intron, which contains the matK gene. Table 2. The basic chloroplast genome information of seven Dactylicapnos speciesCategory of genesGroup of genesName of genesPhotosynthesis related genesATP synthaseatpA,atpB,atpE,atpF*,atpH,atpINADH-dehydrogenasendhA**,ndhB,ndhC,ndhD,ndhE,ndhF,ndhG,ndhH,ndhI,ndhJ,ndhKCytochrome b/f complexpetA,petB*,petD*,petG,petL,petNPhotosystem IpsaA,psaB,psaC,psaI,psaJPhotosystem IIpsbA,psbB,psbC,psbD,psbE,psbF,psbH,psbI,psbJ,psbK,psbL,psbM,psbN,psbT,psbZRubiscorbcLSelf-replicationDNA-dependent RNA polymeraserpoA,rpoB,rpoC1*,rpoC2Large subunit of ribosomerpl2**,rpl14,rpl16,rpl20,rpl22,rpl23,rpl32,rpl33,rpl36Small subunit of ribosomerps2,rps3,rps4,rps7,rps8,rps11,rps12*,rps14,rps15,rps16*,rps18,rps19Ribosomal RNAsrrn5,rrn4.5,rrn16,rrn23Transfer RNAstrnA-UGC*,trnC-GCA,trnD-GUC,trnE-UUC,trnF-GAA,trnfM-CAU,trnG-GCC**trnG-UCC*,trnH-GUG,trnI-CAU,trnI-GAU*,trnK-UUU*,trnL-CAA,trnL-UAA*,trnL-UAG,trnM-CAU,trnN-GUU,trnP-UGG,trnQ-UUG,trnR-ACG,trnR-UCU,trnS-GCU,trnS-GGA,trnS-UGA,trnT-GGU,trnT-UGU,trnV-GAC,trnV-UAC*,trnW-CCA,trnY-GUAOther genesMaturasematKAcetyl-CoA-carboxylaseaccDC-type cytochromesynthesisccsAEnvelope membrane proteincemAATP-dependent proteaseclpPTranslational initiation factorinfAUnknownfunctional genesConserved hypothetical open reading framesycf1,ycf2,ycf3,ycf4,ycf15,ycf68Intron-containing genes are marked by asterisks (*), *gene with one intron; **gene with two introns
Repeat sequence analysis
The studied cp genomes contained 546–878 dispersed repeats, including 360–467 forward repeats (F), 181–410 palindromic repeats (P), 3–23 complement repeats (C), and 1–20 reverse repeats (R), but some Dactylicapnos species do not have complement repeats and reverse repeats (Fig. 2A). Forward repeat was the most universal type, and most dispersed repeats were distributed in two-copy inverted repeat (IR) and large single-copy region (LSC) (Fig. 2B).Fig. 2. Dispersed repeated sequences analyses in cp genomes for seven Dactylicapnos species. A Statistics of four types of dispersed repeats sequences in seven cp genomes; **B **Distribution of dispersed repeats sequences in seven cp genomes
The number of the tandem repeats ranged in the studied cp genomes from 54 to 72. Dactylicapnos torulosa (Hook.f. & Thomson) Hutch. had the most tandem repeats and Dactylicapnos macrocapnos Hutch. had the smallest (Fig. 3A). There were 4 cases of distribution of tandem repeated in the region, distributed in IRa/LSC region, IRb/LSC region, LSC region, SSC/LSC region, most of the tandem repeats are distributed in LSC region (Fig. 3B).Fig. 3. Tandem repeated sequence analyses for seven Dactylicapnos species. A Number of tandem repeats sequences in seven cp genomes; **B **Distribution of tandem repeats sequences in seven cp genomes
Simple sequence repeats (SSRs) analyses
The SSRs were mainly distributed in the LSC region of Dactylicapnos species (Fig. 4A). A total of 327 SSRs were detected in the seven cp genomes, and the number of SSRs ranges from 37 (Dactylicapnos roylei Hutch.) to 60 (D. grandifoliolata), which had the largest number of mononucleotides (35–47), dinucleotides (2–9), trinucleotides (2), hexanucleotides (2), but some Dactylicapnos species did not have trinucleotides and hexanucleotides (Fig. 4B). These SSRs were dominated by mononucleotides (A/T) n. (Fig. 4C), suggesting that the base composition of SSRs is biased toward A/T base.Fig. 4SSRs analyses for seven Dactylicapnos species. A The number of SSRs in LSC, SSC and IRs in seven cp genomes; **B **Number of different SSRs types; **C **Frequency of identified SSRs in different repeat class types
Codon usage analysis
In total, 64 types of codons encoding 20 amino acids were detected, including three termination codons, UAA(), UAG() and UGA(*). The number of codons ranged from 22,187 to 23,325, with the highest number of codons found in D. schneideri, and the lowest number of codons found in Dactylicapnos lichiangensis (Fedde) Hand.-Mazz..
Relative synonymous codon usage (RSCU) values reflect a relationship between the number of actual codon emergence and the number of anticipated codon emergence [22], so that if the RSCU > 1, this mean that the condon has the strong preference. The RCUS calculated from 80 common CDS of the cp genomes of the studied species showed that all protein-coding sequence, 31 codons have RSCU > 1 (strong preference), 31 codons have RSCU < 1 (low preference), Methionine (Met) and threonine (Thr) have no bias (RSCU = 1). (Fig. 5).Fig. 5. The RSCU analysis of 80 common CDS in the chloroplast genomes of Dactylicapnos
IR contraction and expansion
There were differences in the boundary regions of the studied species. In D. macrocapnos, D. scandens, D. schneideri, D. torulosa, and D. lichiangensis, rpl23 was 160–240 bp to the left of the LSC/IRb boundary and trnI was 152–432 bp to the right of the LSC/IRb. The ndhA gene of D. macrocapnos and D. scandens covered the junction of SSC/IRa showed different sizes with 2183 bp and 2184 bp, extending into IRa by 1097 bp and SSC region by 1086 bp and 1087 bp. The ndhI gene of D. grandifoliolata also covered the junction of SSC/IRa, extending into IRa by 333 bp and SSC region by 162 bp. The gene trnH of D. schneideri and D. grandifoliolata was distributed on the left side of the border of IRa/LSC, and the gene trnH of the other species was distributed to the right of the IRa/LSC junction, with an interval of 38–127 bp from the border to the gene (Fig. 6).Fig. 6. Comparison of LSC, IR, and SSC junction positions among seven Dactylicapnos species in cp genomes. JLB denotes the LSC/IRb junction,JSB denotes the SSC/IRb junction, JSA denotes the SSC/IRa junction, and JLA denotes the LSC/IRa junction
Similarity analysis and synteny analysis
Analysis of the level of divergence among the studied species sequences, with D. roylei as a reference, done by mVISTA revealed that the bulk of among sequence variation is located in non-coding intergenic regions and that there were apparent deletions between the coding genes rps3–rpl2 of D. lichiangensis, D. torulosa and D. grandifoliolata (Fig. 7).Fig. 7. Visualization of genome alignment of the chloroplast genomes of seven Dactylicapnos species using D. roylei as a reference by mVISTA. The x-axis represents the coordinate in the chloroplast genome. The Y-axis represents different species, and sequence similarity of aligned regions is displayed as horizontal bars, which expresses as a percentage within 50–100%
The synteny analysis revealed some genomic rearrangements and inversions in the seven cp genomes. Due to the expansion of IR region, the nucleotide sequences of in cp genomes of D. schneideri and D. grandifiliolata was rearranged, and some single copy regions of D. torulosa, D. macrocapnos, D. scandens and D. grandifoliolata were inverted (Fig. 8).Fig. 8. Synteny analysis for seven Dactylicapnos species in chloroplast genomes
Phylogenetic analysis
The maximum likelihood (ML) and bayesian inference (BI) phylogenetic trees (Fig. 9) were constructed using 78 common CDS of the cp genomes of 10 Fumarioideae species, including seven newly sequenced Dactylicapnos species and three outgroups including Lamprocapnos spectabilis (L.) Fukuhara, Corydalis adunca Maxim. and Corydalis edulis Maxim.. The ML and BI methods yielded identical tree topologies with full support for each node (MLBS = 100% and BIPP = 1). The genus was found to be monophyletic and the species of Dactylicapnos formed three distinct clades. The clade consistsing of D. schneideri, and D. grandifoliolata were sister to the rest of Dactylicapnos. D. schneideri formed an independent informal group, and was clustered together with D. grandifoliolata of sect. Pogonosperma. Section Dactylicapnos including D. scandens and D. macrocapnos were sister to sect. Minicalcara including D. lichiangensis, D. roylei and D. torulosa with full support (MLBS = 100% and BIPP = 1).Fig. 9. Maximum likelihood and Bayesian inference phylogeny of Dactylicapnos based on 78 common CDSs of 10 chloroplast genomes. From left to right, Numbers above branches indicate Bayesian posterior probability [BIPP], and Maximum Likelihood bootstrap support [MLBS], respectively. (A, D. macrocapnos; B, D. torulosa; C, D. grandifoliolata; D, D. schneideri)
Discussion
Structure and comparative analysis of Dactylicapnos species
Comparative analysis of cp genomes has been widely used in many plant taxa [23]. In this study, the cp genome of seven Dactylicapnos species were first sequenced, it is also the first time to explore Dactylicapnos species from the molecular analysis. As in the most angiosperms, the cp genome of Dactylicapnos has a typical quadripartite structure [14] but is very long 172,322–176,370 bp being one of the largest cp genomes sequenced to date [24], and the genomic size of the SSC region ranges from 9303 bp to 26,089 bp, with a number difference of about 16 kb, indicating the weakest conservatism and stability. The seven cp genomes are similar in structure, and had from 133 to 140 genes, which indicates that Dactylicapnos cp genomes are structurally conserved and rich in genetic information, which is a reliable molecular material for phylogenetic studies.
The highly conservative IR region is thought to play an important role in stabilizing the chloroplast genome structure [25]. Expansion and contraction of the IR region is a common phenomenon in plant evolutionary history responsible for cp genome length variation [26], which affects the cp genome’s rate of evolution [27, 28], examples are early-diverging eudicots [29, 30] and Apiales [31]. There have been many research about the expansion and contraction of the IR region, and the expansion mechanism of the IR region, the major viewpoint is that minor and apparently random IR expansion may be caused by gene conversion, and larger IR expansion may be achieved through double-strand DNA breaks and subsequent repair mechanism [32, 33], and the contraction mechanism of the IR region is also assumed to be the double-strand DNA breaks and subsequent repair mechanism [34]. In the present study, the IR region has significant expansion or contraction, forming a variety of boundary genes, and the seven cp genomes can be divided into three types according to their variability, which are consistent with the clustering results of the phylogenetic analysis. The gene location information in the boundary region can reveal the phylogenetic relationships between species to some extent [35]. In addition, as the expansion of the IR region at the LSC-IRb boundary, the trnH gene of D. schneideri and D. grandifoliolata entered the IR region leading to genomic rearrangement of these two sequences and the trnH gene becames gene with two copies. The chloroplast genome has multiple copies in the cell and has sufficient interspecific differentiation [35], chloroplast genome sequences for species identification is one of the best methods at present [36], while the cp genome of Dactylicapnos species have significant differences in expansion and contraction, and there are obvious differences in the size of the LSC, SSC, and IR regions of seven cp genomes, suggesting that Dactylicapnos species have a high degree of interspecies differentiation, which can be utilized to adequately demonstrate the phylogenetic relationships between Dactylicapnos species through the cp genomes.
Repeat sequences and SSRs
The plastid genome contains many oligonucleotide repeat sequences that are considered biomarkers of mutational hotspots [37, 38]. Repeat sequences have an important position in genome rearrangements and an important molecular marker in phylogenetic studies [39, 40]. In this present study, four different types of repeat sequences were detected, with the highest number of forward repeats (F) and the lowest number of complement repeats (C). The composition of different types of repeat sequences affects the inheritance and evolution of species [41]. There are small differences in the number and type of repeats among closely related species, both D. schneideri and D. grandifoliolata have four types of repetitive sequences with high similarity in type and number, inferring that the two species may have similarities in genetics and evolution [42]. SSRs are repeated DNA motifs with 1–6 nucleotides and have high polymorphism rates at the species level, have been extensively investigated in population genetics, phylogeography and variety identification [43, 44]. In this study, we found that the types of SSRs in seven cp genomes were found to be essentially the same, but the number of sequences contained in each type was different. Most SSRs loci were distributed in LSC region, with size ranging from 10–125 bp. The mononucleotide (A/T) was the highest proportion in the cp genomes of seven Dactylicapnos species, were found in all species. SSRs polymorphisms are repeat length polymorphisms caused by elongation or shortening of repeat units [45], it is a common molecular tool used to study the evolution of species. In the Camellia [46] and Triticum [47] plant, genetic diversity analysis was performed by amplifying SSRs primers, which led to the construction of genetic evolutionary relationships among species. The large number of SSRs detected in this research can be used as potential molecular markers for subsequent studies of Dactylicapnos species and also provide a theoretical basis for interspecific identification.
Codon usage analysis
The codons that encode the same amino acid are called synonymous codons [48]. In the process of species evolution, synonymous codons are not only associated with nature selection, mutation and genetic drift [49, 50], but also affected by factors such as genome size [51], tRNA abundance [52, 53] and gene expression levels [54], resulting in the genetic codes of different species tend to use one of several synonymous codons, called codon usage bias, which a common feature of eukaryotic genomes and is essential for the regulation of gene expression [55]. The results of the codon usage analysis showed that 31 codons had RSCU values > 1, indicating a codon bias in the amino acids, but unlike other dicotyledons plants [56], these 31 codons of Dactylicapnos do not prefer to end in A/U, It is possible that different levels of evolutionary pressures in Dactylicapnos species have biased the use of codons in this chloroplast genome, but the mechanisms involved need to be further explored [57, 58].
Phylogenetic analysis
The cp genome sequences have been successfully used to reveal phylogenetic relationships [59]. However, due to the different degree of gene rearrangement and inversion in Dactylicapnos, there are significant differences in gene order between sequences, and reliable phylogenetic relationships could not be established using the whole chloroplast genome. The analysis of 78 common CDS from the cp genomes of seven Dactylicapnos species and three outgroups showed that seven Dactylicapnos species were divided into three major clades with full support. Recent classification of Dactylicapnos based on morphology divided the genus into three sections and two informal groups [5]. Our study covered all three sections and one informal group, and our results basically clarified the infrageneric relationships between these three sections and one informal group. The first separated clade includes D. schneideri of an independent informal group sensu Lidén and Pathak [5] and D. grandifoliolata of sect. Pogonosperma sensu Lidén and Pathak [5]. The cp genomes of both D. schneideri and D. grandifoliolata had genomic rearrangements and contracted in the IR regions, and were clustered into the same clade, indicationg their close genetic relationship. The other two clades correspond to the two sections sensu Lidén and Pathak [5], sect. Dactylicapnos and sect. Pogonosperma, respectively. There were differences in the number of genes and GC content of these two clades, and there were also obvious differences in morphological characteristics. D. macrocapnos and D. scandens which were perennial plants with cylindrical stems and small flat globular elaiosomes [2], while D. torulosa, D. roylei and D. lichiangensis were all annual plants with winged-ridged stems and irregular mass elaiosomes [3]. D. scandens and D. macrocapnos of sect. Dactylicapnos were clustered together with full support, and D. lichiangensis, D. roylei and D. torulosa of sect. Minicalcara were also clustered together, so we confirmed sect. Dactylicapnos and sect. Minicalcara based on the plastome phylogenomics.
Conclusion
The cp genome of Dactylicapnos species had a typical tetrad structure and high sequence conservation. A total of 133–140 genes were annotated in the seven Dactylicapnos species, and a large number of repeat sequences and SSRs detected were important molecular markers in population genetics and phylogenetics. Expansion of IR regions and genomic rearrangements revealed by comparative genomic analysis played an important role in the evolution of Dactylicapnos species, and showed that the cp genomes of the D. macrocapnos and D. scandens were closer in structural variation, the D. schneideri was similar to and D. grandifoliolata, while the D. torulosa, D. lichiangensis and D. roylei were more consistent, which supported the results of phylogenetic analyses that categorized the seven species of Dactylicapnos into three clades. In addition, the most comprehensive and robust phylogeny covering all three sections and one informal group of Dactylicapnos based on cp genomes was reconstructed to basically clarify infrageneric relationships for the first time. Phylogenetic analysis showed that seven species separated into three major evolutionary clades, which suggested that this genus should be divided into three sections. The novel genomic resources provided here will aid future study in development of medicine resources, infrageneric classification, character evolution, diversification and biogeography. It also showed that the structural information and variation of chloroplast genomes were important for phylogenetic analysis, providing strong evidence for a deeper understanding of phylogenetic relationships and evolution among species.
Materials and methods
Plant material, DNA extraction and sequencing
Most of the material of Dactylicapnos species were fresh leaves collected in the field and dried with silica gel, and a few materials were obtained from the herbarium of KUN (Herbarium, Kunming Institute of Botany, CAS) and PE (Herbarium, Institute of Botany,CAS) (Table 3). The DNA extraction, library preparation and shallow sequencing were performed by Novogene, and the library was sequenced on the Illumina Hiseq 4000 platform with 150 bp paired-end reads. For the herbarium specimens, the method of Zeng et al. [60] was adopted for sequencing and library construction. Table 3. Species Collection InformationSpeciesHerbariumCollection numberDeterminavitLocalitycoordinate information (Lat, Lon)GenBank accessionD. macrocapnosHerbarium, Institute of Botany, CAS (PE)01029Juntong Chen, Shunquan YangNyalam, Xizang, China27.97°, 85.96°OR589107D. scandensHerbarium, Kunming Institute of Botany, CAS (KUN)LuJL118Juntong Chen, Shunquan YangPingBian, Wenshan Zhuang and Miao Autonomous Prefecture, Yunnan, China23.13°, 104.78°OR568573D. schneideriHerbarium, Kunming Institute of Botany, CAS (KUN)10,566Juntong Chen, Shunquan YangYanyuan, Liangshan Yi Autonomous Prefecture, Sichuan, China27.56°,101.75°OR589106D. grandifoliolataHerbarium, Kunming Institute of Botany, CAS (KUN)Deng-15218Juntong Chen, Shunquan YangYadong, Rikaze, Xizang, China27.24°, 89.02°OR589105D. torulosaHerbarium, Kunming Institute of Botany, CAS (KUN)SunH-07ZX-3200Juntong Chen, Shunquan YangYulong, Lijiang, Yunnan, China26.79°, 99.64°OR589104D. lichiangensisHerbarium, Kunming Institute of Botany, CAS (KUN)SunH-07ZX-3234Juntong Chen, Shunquan YangYulong, Lijiang, Yunnan, China26.78°, 99.67°OR589103D. royleiHerbarium, Kunming Institute of Botany, CAS (KUN)38,434Juntong Chen, Shunquan YangXiaojin, Sichuan, China30.99°, 102.69°OR568572
Chloroplast genome assembly, annotation and codon usage
De novo assembly of the cp genome was carried out using GetOrganelle 1.7.6.1 [61]. We used the genome annotator PGA [62] to annotate the sequences that have been assembled into loops using the Lamprocapnos spectabilis (NC_039756) as the reference, and manually correct the position of the start and stop codons and the boundary between the exons and introns with Geneious Prime 2023.0.4 [63]. Finally, the physical maps of cp genome were created by using OrganellarGenomeDRAW (https://chlorobox.mpimp-golm.mpg.de/OGDraw.html) [64]. The RSCU was the ratio of the frequency of a specific codon to the expected frequency of that codon, which was obtained by Genepioneer platform, and plotted the heatmap of RSCU values with TBtools 1.116 [65].
Analysis of repeat sequences and SSRs
Repeat sequences in the cp genome were detected by REPuter [66], including forward, palindromic, reverse and complement repeats, the parameters were set with minimum repeat size 30 bp, and an hamming distance of 3. And exploring tandem repeats of cp genome by the Tandem Repeat Finder [67]. The simple sequence repeats (SSRs) were identified by using MISA online tool (https://webblast.ipk-gatersleben.de/misa/) [68], and the repeat thresholds for mononucleotide, dinucleotide, trinucleotide, tetrtanucleotide, pentanucleotide and hexanucleotide SSRs were 10, 6, 5, 5, 5, 5, respectively.
Comparative genomic analyses
The online program IRscope (https://irscope.shinyapps.io/irapp/) [69] was used to study the expansion and contraction of the IR region in the cp genome sequence of Dactylicapnos species. The genome comparison of the seven Dactylicapnos species in the cp genomes was analyzed by the mVISTA (https://genome.lbl.gov/vista/index.shtml) [70] program with the Shuffle-LAGAN mode, and the synteny analysis of cp genome was performed with Mauve [71].
Phylogenetic analysis
Phylogenetic analysis was performed based on 78 common CDS of the cp genomes of 10 Fumarioideae species, including seven Dactylicapnos cp genomes and three closely related species (Lamprocapnos spectabilis, Corydalis adunca and Corydalis edulis). These three species were selected as outgroups based on previous phylogenetic results[10–12], and these three plastomes were downloaded from GenBank. All sequences were aligned using MAFFT and maximum likelihood (ML) analysis was performed by RAxML-8.2.12 on CIPRES (https://www.phylo.org/portal2/) website with the GTRGAMM model, and 1000 bootstrap replicates. The best-fit model GTR + I + G was selected by AIC (Akaike Information Criterion) with jModelTest 2.1.10 [72], and the Bayesian inference (BI) analyses were conducted by MrBayes-3.2.7 on CIPRES website, with the settings: four MCMC simulations were run simultaneously and sampled every 1,000 generations for a total of two million generations, the first 25% of trees were discarded as burn-in.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Wu ZY Zhang X Su ZY Flora Reipublicae Popularis Sinicae Beijing Science Press.1999328893
- 2Lidén M. Three new species of Dactylicapnos (Fumariaceae) and a synopsis of the D. macrocapnos complex. Nord J Bot. 2010;28(6):656–60.
- 3Lidén M, Pathak MK. Studies in Dactylicapnos (Papaveraceae–Fumarioideae) part II Revision of Dactylicapnos sect. Pogonosperma sect. nov with D.arunachalensis sp nov. Nord J Bot. 2014;32(2):176–84.
- 4Wang FH Hu X Chen HL Ma JP Wang JX Hou AJ Alkaloids from Dactylicapnos scandens Hutch China J Chin Materia Med 200934162057205919938545 · pubmed ↗
- 5Guo CC. The metabolism and pharmacokinetics of isocorydine and protopine in Dactylicapnos scandens. Zhejiang University; 2013. p. 2–7.
- 6Wang B Zhao YJ Zhao YL Liu YP Li XN Zhang HB Luo XD Exploring aporphine as anti-inflammatory and analgesic lead from Dactylicapnos scandens Org Lett 201922125726010.1021/acs.orglett.9b 0425231860319 · doi ↗ · pubmed ↗
- 7Lidén M Fukuhara T Rylander J Oxelman B Phylogeny and classification of Fumariaceae, with emphasis on Dicentra sl, based on the plastid gene rps 16 intron Plant Syst Evol 199720641142010.1007/BF 00987960 · doi ↗
- 8Perez-Gutierrez MA Romero-Garcia AT Salinas MJ Blanca G Fernandez MC Suarez-Santiago VN Phylogeny of the tribe Fumarieae (Papaveraceae s.l.) based on chloroplast and nuclear DNA sequences: evolutionary and biogeographic implications American Journal Botany.201299351752810.3732/ajb.110037422334448 · doi ↗ · pubmed ↗