IFITM5-related (type V) osteogenesis imperfecta with evidence of perinatal involvement: A case report
Valentina Martínez-Montoya, Miguel Angel Fonseca-Sánchez, Gerardo Fabian-Morales, Ramiro Vega-Gamas, Gloria Eugenia Queipo-García, Luis Felipe León-Madero, Luz María Sánchez-Sánchez

TL;DR
A 3-month-old girl with bone fractures and deformities was diagnosed with a rare form of osteogenesis imperfecta linked to the IFITM5 gene, with evidence of prenatal involvement.
Contribution
This case is the first to report prenatal manifestations associated with the IFITM5 c.-14C>T variant in osteogenesis imperfecta.
Findings
The patient exhibited progressively deforming OI traits consistent with IFITM5-related disease.
The c.-14C>T variant was identified, with suspected prenatal bone manifestations.
Prenatal involvement had not been previously reported for this IFITM5 variant.
Abstract
Osteogenesis imperfecta (OI) is a rare hereditary disorder characterized by bone fragility and frequent fractures. While most cases are attributed to variations in collagen-coding genes COL1A1 and COL1A2, other genes such as IFITM5 have also been associated with the disease, accounting for up to 5 % of cases. Here, we report a case of a 3-month-old female with a femur fracture and limb deformity. X-rays revealed evidence of osteopenia and previous fractures in the arms, clavicle, ribs, and left limb, alongside prenatal bone deformities detected by ultrasound. Initial clinical evaluation suggested progressively deforming (Sillence's type III) osteogenesis imperfecta (OI). Molecular testing led to the diagnosis of IFITM5-related OI, identifying the c.-14C>T (rs587776916) variant. Although this variant has been previously reported in patients with IFITM5-related OI, prenatal involvement…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
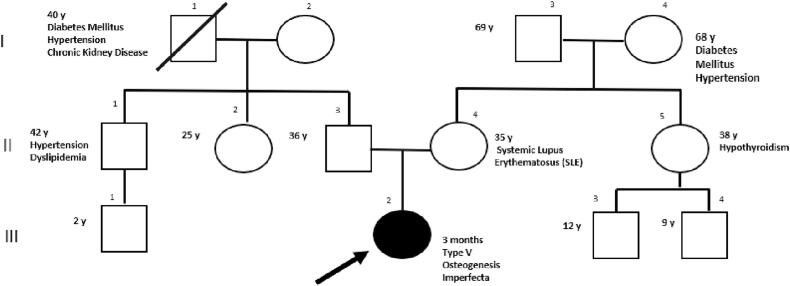 Figure 1
Figure 1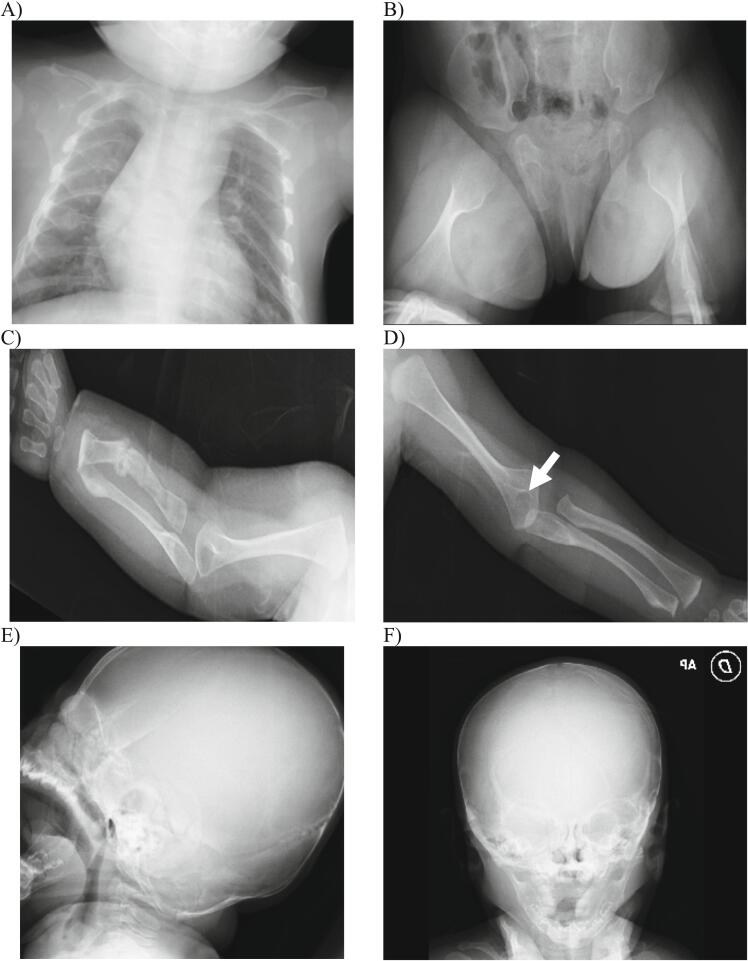 Figure 2
Figure 2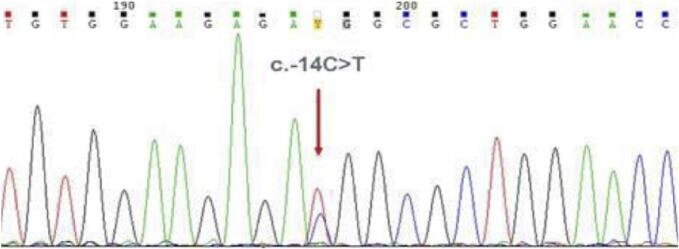 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsConnective tissue disorders research · Bone and Dental Protein Studies · Bone health and treatments
Introduction
1
Osteogenesis imperfecta (OI) is a genetic disorder of connective tissue characterized by abnormalities in collagen synthesis or processing (Subramanian et al., 2023). Individuals with OI often exhibit a heightened susceptibility to bone fractures due to reduced bone mineral density, earning the colloquial term “brittle bone disease.” (Marini et al., 2007) Core features encompass fractures, bone deformities, and short stature. Nevertheless, extra-skeletal manifestations are also common, including dentinogenesis imperfecta, blue sclera, and hearing disorders (Lindahl et al., 2015). Although variants in COL1A1 and COL1A2 genes have traditionally been implicated in up to 90 % of cases of OI, advancements in NGS over the last decade have unveiled a plethora of genes associated with this disease (Lindahl et al., 2015; Unger et al., 2023).
Traditionally, OI has been classified based on clinical and radiological characteristics, with types I - IV associated with the COL1A1 and COL1A2 genes, constituting Sillence's classification, which remains widely utilized. These types are autosomal dominant with blue sclerae (type I), perinatal lethal (type II), progressively deforming (type III), and autosomal dominant with normal sclerae (type IV) (Van Dijk et al., 2010). Additionally, type V was later described as a form of OI characterized by calcification in the intraosseous membranes. Recently, with the discovery of new variants associated with OI, more comprehensive classifications and terminologies have been proposed to reflect the diversity of genes associated with this condition (Table S1) (Homan et al., 2011; Van Dijk et al., 2009; Baldridge et al., 2008; van Dijk et al., 2009; Christiansen et al., 2010; Kelley et al., 2011). As various terminologies and classifications are used interchangeably throughout the literature, we will refer to this disorder in a gene-based manner, addressing Sillence's classification as necessary. Readers are encouraged to refer to the supplementary material for a brief review of terminology and nomenclature.
Type V OI, according to Sillence's classification, is caused by a Single Nucleotide Variant (SNV) in the 5′ untranslated region (5’-UTR) of IFITM5 gene, which anticipates the start codon and elongates the N-terminal region of the bone-restricted IFITM protein (BRIL), also known as IFM5 for the addition of five new amino acids to the wild-type protein (Hanagata, 2016). In addition to variants in COL1A1 and COL1A2, variants in IFITM5 have been linked to an autosomal dominant inheritance pattern (Forlino and Marini, 2016). Roughly 5 % of all OI cases are attributed to variants in IFITM5 (Jovanovic et al., 2022). The phenotype of IFITM5-related OI differs from others due to the presence of calcification of the interosseous membrane and the formation of hyperplastic callus. However, this type has a broader phenotypic spectrum than others (Glorieux et al., 2000; Rauch et al., 2013).
In this case report, we present an infant with IFITM5-related OI exhibiting a classic severe presentation and no familial history of bone pathology. The patient displayed clinical features consistent with progressively deforming OI (type III according to Sillence's classification) with perinatal manifestations. Sequencing of IFITM5 uncovered a C>T de novo occurring variant in the c.-14 position of the 5’UTR of exon 1, corresponding to rs587776916.
Subject and methods
2
Patient report
2.1
The proband, a 3-month-old female patient, was referred to the genetics clinic after experiencing spontaneous fractures. She was the first pregnancy of nonconsanguineous parents of mexican ancestry. Her mother was 34 years old and her father was 37 years old at the patient's birth. Her mother had a clinical history of systemic lupus erythematosus diagnosed at the age of 26, which had been managed with chloroquine, prednisone, and azathioprine. She had been in remission without requiring medical treatment for the past 5 years. Extended familial history included a paternal uncle with hypertension and mixed dyslipidemia, a maternal aunt with hypothyroidism, a paternal grandfather with chronic kidney disease, diabetes, and hypertension (deceased at 40 years of age), a paternal grandmother with hypertension, and a maternal grandmother with hypertension and diabetes. There was no known familial history of bone pathology (Fig. 1).Fig. 1. Proband III 2. IFITM5-related osteogenesis imperfecta with prenatal involvement. De novo gene variant in IFITM5. No familial history of bone pathology. Parents are not carriers of the IFITM5 variant.Fig. 1
Her mother received adequate prenatal care, including the intake of multivitamins, iron, and folic acid throughout her pregnancy. However, during week 33, an ultrasound examination indicated moderate oligohydramnios (amniotic fluid index: 8.2 cm). In response, the attending physician advised physical rest and regular fetal monitoring. Subsequent ultrasound scans showed areas of shortening and curvature in the humeri compared to previous images. The patient was delivered via cesarean section at 37 weeks of gestation, weighing 2535 g and measuring 45 cm at birth. Extended neonatal screening and auditory testing yielded normal results, and no apparent morphological abnormalities were observed. Further radiological investigations were not deemed necessary.
The patient exhibited irritability and daily crying as reported by the mother. At 3 months of age, an increased volume in the left lower limb above the knee and uncontrollable crying were also observed. The patient was evaluated by a pediatrician, who identified multiple old bone fractures in the upper limbs and left femur. Subsequently, immobilization with a cast was applied to the left leg. The patient was then referred to the genetics clinic, where low weight (5.5 kg, −1.03SD) and height (55 cm, −2.98 SD) were noted. Additionally, the patient exhibited positional plagiocephaly predominantly affecting the right temporoparietal region, blue-gray sclerae, hypertelorism, triangular face, and ulnar deformity of the right forearm. Vital signs were within normal range, and a renal ultrasound revealed no abnormalities. The results of laboratory tests and clinical findings are displayed in Table 1.Table 1. Clinical features and lab test results.Table 1. Clinical findings
-
•Positional plagiocephaly predominantly at the right temporoparietal region
-
•Blue-grayish sclera
-
•Anterior auricular pits in the right auricle lobe
-
•Hypertelorism, triangular face
-
•Ulnar deviation of right forearm
-
•Low weight (5.5 kg, −1.03SD) and height (55 cm, −2.98 SD) Lab workout (3 months age)ValueNormal value or intervalCalcium: 10.3 mg/dL9–11 mg/dLPhosphorous: 5.9 mg/dL3.7–6.5 mg/dLMagnesium: 2.1 mg/dL1.5–2.2 mg/dLParathormone: 55.48 pg/mL15–65 pg/mL25-OH cholecalciferol (vitamin D): 32.14 ng/ mL30–100 ng/mLALP: 935 U/L< 449 U/LAST: 57 U/L< 32 U/LGGT: 24 U/L< 33 U/LCreatinine: 0.21 mg/dLNABUN: 6.5 mg/dLNAALP = alkaline phosphatase; AST = aspartate transaminase; BUN = blood urea nitrogen; GGT = gamma-glutamyl transferase; NA = not available.
Radiological tests revealed evidence of overall osteopenia, particularly affecting the long bones, with luminescent bands near the growth cartilages observed in the wrist and left femur. No calcification of the interosseous membranes was identified. Furthermore, an unresolved distal fracture in the left femur and a non-displaced fracture in the left forearm with a bone callus were observed. The presence of bone callus formation observed in X-rays at 3 months old, coupled with ultrasound findings in the third trimester indicating shortened and curved limbs, led to the conclusion that the fractures likely occurred during the pregnancy. Multiple images of old fractures are depicted in Fig. 2A-F.Fig. 2A) Chest X-ray showed callus formation in the 4th, 5th, and 6th left costal arches, as well as the 6th and 7th right costal arches and left clavicle, indicating probable fractures occurring during the prenatal stage. B) Pelvic X-ray revealed a distal metaphyseal fracture of the left femur, angled dorsally at 40°, without involvement of the joint. Pathological widening of the metaphysis was noted, along with overall osteopenia. C) X-ray of the upper right extremity displayed an old diaphyseal fracture of the forearm with bone callus formation. Additionally, a new fracture of the distal radial metaphysis was observed, with the patient wearing a splint at the time of evaluation. D) X-ray of the upper left extremity revealed an old, non-displaced, and non-angulated distal metaphyseal fracture of the humerus. E and F) Cranial X-rays revealed overall demineralization in the diploe, along with Wormian bones in the lambdoid suture.Fig. 2
Based on the phenotype and imaging evidence, progressively deforming (Sillence's type III) OI was suspected. Sequencing of a 24-gene panel associated with OI was requested in order to confirm the diagnosis.
Next-generation sequencing
2.2
Parental informed consent was obtained, followed by the extraction and analysis of genomic DNA. Briefly, DNA was extracted with a commercial kit (GeneAll® Biotechnology, Seoul, Korea) and targeted next-generation sequencing (NGS) was performed in an Illumina NextSeq System (Illumina, CA, USA; Agilent Technologies, CA, USA). Variants were evaluated in the exonic and flanking intronic regions (±8 bp) of the probed genes: ALPL, B3GALT6, BMP1, COL1A1, COL1A2, CREB3L1, CRTAP, FKBP10, IFITM5, LRP5, NOTCH2, P3H1, P4HB, PLOD2, PLS3, PPIB, SEC24D, SERPINF1, SERPINH1, SP7, SPARC, TAPT1, TMEM38B, and WNT1. Genome Analysis Toolkit (GATK, Broad Institute, MA, USA) was used for alignment against a reference genome (hg19). 96.4 % of targeted regions were covered at a sequencing depth no <10×, with 94.7 % covered at a sequencing depth no <20×. Results were validated by Sanger sequencing for the proband and parents. Briefly, PCR reactions were conducted using gDNA at a concentration of 1 ng/μL with GoTaq® Flexi DNA polymerase (Promega Corp., WI, USA) following manufacturer's protocol. Primers used for the IFITM5 gene were: 5′-CAGATGGGATGTCTGTCAGGAG-3′ and 5′-CAGATTCAGGTAGAGGGTGCTG-3. End products were verified by agarose gel electrophoresis.
Patient consent statement
2.3
The parents of the patient provided consent for the publication of this clinical case. Following a comprehensive explanation of the publication process, the parents signed an informed consent form specifically for publication purposes.
Statistical analysis
2.4
No comparative statistical analysis was conducted as this was a descriptive study.
Results
3
NGS revealed the presence of a heterozygous variant NM_001025295.3: c.-14C>T in the 5' UTR of exon 1 in the IFITM5 gene corresponding to rs587776916. This variant has been previously associated with IFITM5-related OI (corresponding to Sillence's Type V OI, MIM 610967) which exhibits an autosomal dominant inheritance pattern. In accordance with the American College of Medical Genetics and Genomics (ACMG) criteria this variant was classified as probably pathogenic (PM2, PP5). Sanger sequencing of proband confirmed this result (Fig. 3), subsequent analysis of parental DNA revealed a de novo occurrence.Fig. 3. The electropherogram of exon 1 of the IFITM5 gene demonstrates the substitution of cytosine to thymine (c.-14C>T) in the 5’-UTR region of the gene. This gene variant results in the creation of an alternative upstream start codon within the ORF of IFITM5, leading to the production of five new amino acids (Met-Ala-Leu-Glu-Pro) in the N-terminal region of the interferon-induced transmembrane protein 5.Fig. 3
Discussion
4
IFITM5 gene belongs to the interferon-induced transmembrane gene family. It is located at the 11p15.5 locus and encodes a protein consisting of 132 amino acids. This protein features two transmembrane domains, N- and C-terminal, along with an intracellular loop. Unlike other genes of the IFITM family, IFITM5 lacks interferon response elements and is not induced by interferons (Cho et al., 2012). The pathogenic heterozygous variant c.-14C>T (rs587776916) anticipates a start codon which is in-frame with the ORF of the IFITM5 gene, leading to the addition of 5 new amino acids to the IFITM or BRIL (bone-restricted IFITM-like) proteins. This protein appears to have a crucial role during bone mineralization. The c.-14C>T (rs587776916) variant has been previously reported in the literature as pathogenic associated with type V (according to Sillence's classification) OI (Forlino and Marini, 2016; Rauch et al., 2013; Cho et al., 2012; Grover et al., 2013; Semler et al., 2012).
Murine models of IFITM5-related OI have demonstrated a slow rate of mineralization in utero and abnormal rib cage formation and long bone deformities and fractures. Notably, gene expression levels in osteoblasts of mutant mice were reduced compared to wild-type mice. Additionally, a reduction in bone mineralization was observed. These results highlight the influence of the c.-14C>T variant on bone formation, contrasting with previous studies that did not observe significant bone involvement in murine models. In an IFITM5-related OI mouse model carrying the c.-14C>T variant, perinatal lethality was observed, possibly due to respiratory failure resulting from abnormal rib cage formation. Consequently, it was not possible to determine whether the formation of hyperplastic callus and calcification of the interosseous membrane occurs in the prenatal or postnatal stage (Hanagata, 2016).
Type V (according to Sillence's classification) OI was originally characterized as a distinct OI subtype encompassing hyperplastic callus formation, interosseous membrane calcifications and radioopaque metaphyseal bands in absence of COL1A1 or COL1A2 alterations (Glorieux et al., 2000; Rauch et al., 2013). However, these features are not consistent among the patients diagnosed with Type V OI (according to Sillence's classification). For instance, up to 35 % of patients do not exhibit hyperplastic callus. Moreover, phenotype-modifying genes have been proposed as an underlying mechanism of the phenotypic variability observed in these patients (Semler et al., 2012).
This case report portrays a 3-month-old patient with molecular diagnosis of IFITM5-related OI caused by a de novo c.-14C>T variant (rs587776916). The patient exhibited an intermediate phenotype between those previously described in the literature. Prenatal evidence of upper limb shortening was documented at 35 weeks of gestation. At 3 months of age, a spontaneous fracture in the left femur prompted radiological studies which revealed old fractures and hyperplastic bone calluses, without evidence of calcification of the interosseous membranes of the arms or radial head dislocation. Additionally, the patient exhibited blue-gray discoloration of the sclera. The biochemical bone panel showed an elevation in alkaline phosphatase (ALP), indicating increased bone turnover or growth. Unfortunately, a urinary excretion test for collagen type I N-telopeptide could not be conducted to confirm this suspicion.
IFITM5-related OI exhibits a wide phenotypic variability. Patients with clinical features resembling progressively deforming or mild to moderate with normal sclerae OI (type III or type IV according to Sillence's classification) have been reported to be identified as having IFITM5-related OI following molecular testing (Grover et al., 2013). This is relevant to our case, as our patient was initially classified as having progressively deforming OI (type III according to Sillence's classification) until molecular testing confirmed the diagnosis of IFITM5-related OI.
It is not uncommon to encounter patients who do not exhibit all the radiological findings associated with this subtype, supporting the hypothesis of variable expressivity (Semler et al., 2012). For instance, Grover et al. documented a case of IFITM5-related OI where the patient lacked radiological evidence of interosseous membrane ossification in the arms and did not exhibit typical clinical features such as prono-supination alterations, radial head dislocation, and radiodense metaphyseal bands associated with this condition (Grover et al., 2013). In this case, prenatal ultrasound revealing upper limb shortening prompted classification as progressively deforming OI (type III according to Sillence's classification) prior to the availability of molecular diagnosis.
Prenatal involvement of patients with IFITM5-related OI has been reported for other variants in this gene. For instance, Gillen Navarro et al. documented a novel variant NM_001025295.3 c.119C>T p.Ser40Leu (rs786201032) in the IFITM5 gene in a patient exhibiting prenatal limb shortening and the absence of typical clinical features of type V OI. This finding highlighted the lack of allelic heterogeneity in IFITM5 and raised the suspicion that different variants in IFITM5 lead to distinct clinical presentations of OI. Additionally, it reported the first case of OI associated with a pathogenic variant in the coding region of exon 1 of IFITM5 with prenatal involvement (Guillén-Navarro et al., 2014). Hoyer-Kuhn et al. reported a similar case involving the same variant reported by Gillen Navarro in a patient with IFITM5-related OI and prenatal involvement (Hoyer-Kuhn et al., 2014). Interestingly, Farber et al. identified the same variant in a 25-year-old woman with severe OI (Farber et al., 2014).
To the best of our knowledge, no cases of IFITM5-related OI with prenatal involvement caused by c.-14C>T (rs587776916) variant had been previously reported in the literature. Our patient did not exhibit ultrasonographic evidence of fractures or skeletal dysplasia during the second trimester of gestation, in contrast to the discovery of upper limb shortening during the third-trimester ultrasound, implying that the condition may have developed during this period. Clinical manifestations indicative of OI were evident in our patient within the first three months of life, suggesting that fractures may have occurred prenatally. These observations raise the suspicion of prenatal manifestations of IFITM5-related OI associated with the c.-14C>T (rs587776916) variant. However, due to the absence of morphological cues at birth, imaging studies were not initially deemed necessary and were consequently not pursued until three months later when the patient's mother sought medical attention for tenderness in the left lower limb. As such, definitive evidence is currently lacking. During the clinical evaluation, no ossification of the interosseous membrane was documented, suggesting that this phenomenon may occur in later stages of the disease's progression.
This case expands the phenotypic spectrum of IFITM5-related OI and strengthens the hypothesis that there may be undiscovered phenotype modifiers linked to pathogenic variants in IFITM5 (Jovanovic et al., 2022; Glorieux et al., 2000; Rauch et al., 2013). Currently, prenatal presentation had been attributed to the c.119C>T p.Ser40Leu variant (rs786201032). However, our case report confirms that c.-14C>T (rs587776916) can also manifest with prenatal presentation, albeit in an advanced stage of fetal development. This observation may be attributed to the dual pathophysiological mechanism observed in several cases of IFITM5-related OI, characterized by severe impairment of bone mineralization and exacerbated, defective bone formation. The c.-14C>T variant has previously been associated with defective bone formation, characterized by the presence of bone callus and membrane ossification. This is attributed to a gain-of-function (GOF) mechanism resulting from protein elongation, which contrasts with bone mineralization alterations associated with other variants (Jovanovic et al., 2022). Future studies may elucidate the mechanisms underlying the variability of clinical manifestations of IFITM5-related OI.
Conclusion
5
IFITM5-related OI encompasses a broad spectrum of clinical manifestations. This case further broadens the phenotypic spectrum associated with the c.-14C>T (rs587776916) variant, presenting with spontaneous fractures at 3 months of age and evidence of multiple previous fractures, likely occurring during the prenatal stage. Additionally, the patient exhibited blue-gray discoloration of the sclera and biochemical abnormalities. These findings expand our understanding of the clinical presentations of IFITM5-related OI, highlighting the possibility of prenatal manifestations associated with the c.-14C>T (rs587776916) variant.
The following is the supplementary data related to this article.Table S1Nosology of osteogenesis imperfecta.Table S1
CRediT authorship contribution statement
Valentina Martínez-Montoya: Writing – review & editing, Writing – original draft, Visualization, Methodology, Investigation, Formal analysis, Conceptualization. Gerardo Fabian-Morales: Resources, Methodology, Investigation, Formal analysis. Miguel Angel Fonseca-Sánchez: Resources, Methodology, Investigation, Formal analysis. Ramiro Vega-Gamas: Resources, Methodology, Investigation, Formal analysis. Gloria Eugenia Queipo-García: Resources, Methodology, Investigation, Formal analysis. Luis Felipe León-Madero: Validation, Supervision, Investigation, Formal analysis. Luz María Sánchez-Sánchez: Writing – review & editing, Validation, Supervision, Project administration, Conceptualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Baldridge D.Schwarze U.Morello R.Lennington J.Bertin T.K.Pace J.M.Pepin M.G.Weis M.Eyre D.R.Walsh J.Lambert D.Green A.Robinson H.Michelson M.Houge G.Lindman C.Martin J.Ward J.Lemyre E.Mitchell J.J.Krakow D.Rimoin D.L.Cohn D.H.Byers P.H.Lee B.CRTAP and LEPRE 1 mutations in recessive osteogenesis imperfecta Hum. Mutat.2920081435144210.1002/humu.2079918566967 PMC 2671575 · doi ↗ · pubmed ↗
- 2Cho T.-J.Lee K.-E.Lee S.-K.Song S.J.Kim K.J.Jeon D.Lee G.Kim H.-N.Lee H.R.Eom H.-H.Lee Z.H.Kim O.-H.Park W.-Y.Park S.S.Ikegawa S.Yoo W.J.Choi I.H.Kim J.-W.A single recurrent mutation in the 5′-UTR of IFITM 5 causes osteogenesis imperfecta type V Am. J. Hum. Genet.91201234334810.1016/j.ajhg.2012.06.00522863190 PMC 3415533 · doi ↗ · pubmed ↗
- 3Christiansen H.E.Schwarze U.Pyott S.M.Al Swaid A.Al Balwi M.Alrasheed S.Pepin M.G.Weis M.A.Eyre D.R.Byers P.H.Homozygosity for a missense mutation in SERPINH 1, which encodes the collagen chaperone protein HSP 47, results in severe recessive osteogenesis imperfecta Am. J. Hum. Genet.86201038939810.1016/j.ajhg.2010.01.03420188343 PMC 2833387 · doi ↗ · pubmed ↗
- 4van Dijk F.S.Nesbitt I.M.Zwikstra E.H.Nikkels P.G.J.Piersma S.R.Fratantoni S.A.Jimenez C.R.Huizer M.Morsman A.C.Cobben J.M.van Roij M.H.H.Elting M.W.Verbeke J.I.M.L.Wijnaendts L.C.D.Shaw N.J.Högler W.Mc Keown C.Sistermans E.A.Dalton A.Meijers-Heijboer H.Pals G.PPIB mutations cause severe osteogenesis imperfecta Am. J. Hum. Genet.85200952152710.1016/j.ajhg.2009.09.00119781681 PMC 2756556 · doi ↗ · pubmed ↗
- 5Farber C.R.Reich A.Barnes A.M.Becerra P.Rauch F.Cabral W.A.Bae A.Quinlan A.Glorieux F.H.Clemens T.L.Marini J.C.A novel IFITM 5 mutation in severe atypical osteogenesis imperfecta type VI impairs osteoblast production of pigment epithelium-derived factor J. Bone Miner. Res.2920141402141110.1002/jbmr.217324519609 PMC 4352343 · doi ↗ · pubmed ↗
- 6Forlino A.Marini J.C.Osteogenesis imperfecta Lancet 38720161657167110.1016/S 0140-6736(15)00728-X 26542481 PMC 7384887 · doi ↗ · pubmed ↗
- 7Glorieux F.H.Rauch F.Plotkin H.Ward L.Travers R.Roughley P.Lalic L.Glorieux D.F.Fassier F.Bishop N.J.Type V osteogenesis imperfecta: a new form of brittle bone disease J. Bone Miner. Res.1520001650165810.1359/JBMR.2000.15.9.165010976985 · doi ↗ · pubmed ↗
- 8Grover M.Campeau P.M.Lietman C.D.Lu J.T.Gibbs R.A.Schlesinger A.E.Lee B.H.Osteogenesis imperfecta without features of type V caused by a mutation in the IFITM 5 gene J. Bone Miner. Res.2820132333233710.1002/jbmr.198323674381 PMC 3800501 · doi ↗ · pubmed ↗