Pathogenic role of super-enhancers as potential therapeutic targets in lung cancer
Zhiyuan Yao, Peng Song, Wenjie Jiao

TL;DR
This paper explores how super-enhancers contribute to lung cancer and could be targeted for new therapies.
Contribution
The paper reviews the role of super-enhancers in lung cancer and highlights their potential as therapeutic targets.
Findings
Super-enhancers are linked to cell identity and cancer progression in lung cancer.
Combination therapies targeting super-enhancers show promise in preclinical models.
Abstract
Lung cancer is still one of the deadliest malignancies today, and most patients with advanced lung cancer pass away from disease progression that is uncontrollable by medications. Super-enhancers (SEs) are large clusters of enhancers in the genome’s non-coding sequences that actively trigger transcription. Although SEs have just been identified over the past 10 years, their intricate structure and crucial role in determining cell identity and promoting tumorigenesis and progression are increasingly coming to light. Here, we review the structural composition of SEs, the auto-regulatory circuits, the control mechanisms of downstream genes and pathways, and the characterization of subgroups classified according to SEs in lung cancer. Additionally, we discuss the therapeutic targets, several small-molecule inhibitors, and available treatment options for SEs in lung cancer. Combination…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
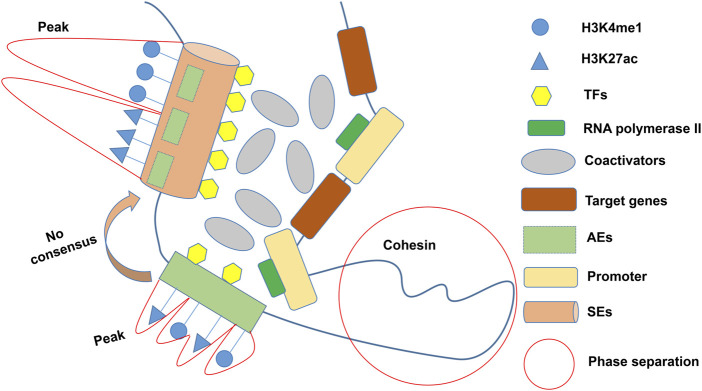 FIGURE 1
FIGURE 1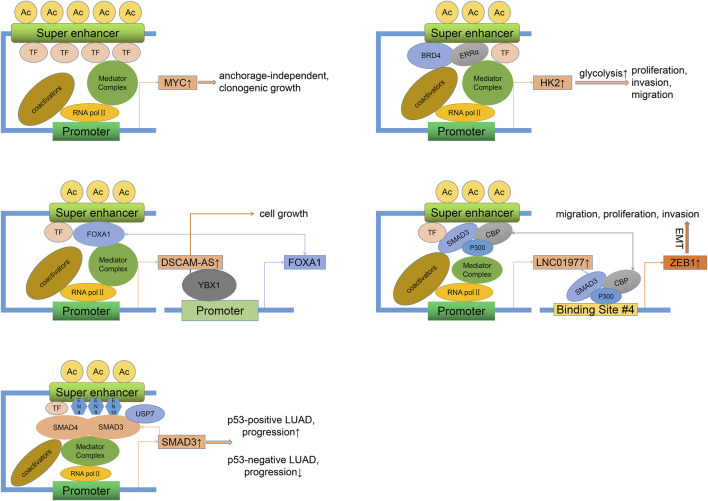 FIGURE 2
FIGURE 2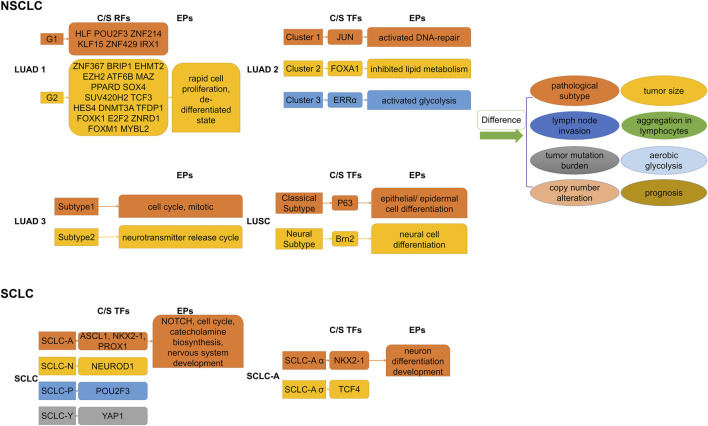 FIGURE 3
FIGURE 3| Target | Compound | Phase | Status | Cancer type | NCT number |
|---|---|---|---|---|---|
| BET/BRD4 | Birabresib | 1 | Terminated | NSCLC, NMC | |
| 1 | Completed ( | NSCLC, NMC | |||
| PLX-2853 | 1 | Completed | SCLC | ||
| Mivebresib | 1 | Completed | NSCLC, SCLC | ||
| Molibresib | 2 | Withdrawn | SCLC | ||
| 1 | Completed ( | SCLC, NMC | |||
| ZEN-3694 | 2 | Recruiting | LUSC | ||
| INCB-057643 | 1/2 | Terminated | NSCLC | ||
| CDK7 | Seliciclib | 2 | Terminated | NSCLC | |
| SY-5609 | 1 | Active, not recruiting | SCLC | ||
| Roniciclib | 2 | Terminated ( | SCLC | ||
| 1/2 | Withdrawn | NSCLC | |||
| 1/2 | Terminated ( | SCLC | |||
| CDK9 | Dinaciclib | 2 | Completed ( | NSCLC | |
| Hydrochloride | 1 | Terminated | NSCLC, SCLC | ||
| PRT-2527 | 1 | Active, not recruiting | NSCLC |
| Agents | Combination | Mechanisms | Cancer type | Malignant features in | References |
|---|---|---|---|---|---|
| QCA570 | Osimertinib | reduction of Mcl-1 | NSCLC | decreased survival, apoptosis, inhibition of formation and colonies growth |
|
| ABBV-075 | Venetoclax | disruption of Bim-Bcl2 complexes, release of Bim | SCLC | apoptosis, tumor regression |
|
| JQ1 | ABT-263 | disruption of Bim-Bcl2 complexes, release of Bim | SCLC | apoptosis |
|
| GSK525762 (I-BET762) | Talazoparib | inhibition of DNA damage response, HR-DSBR process | SCLC | reduced survival and growth |
|
| JQ1 | Paclitaxel | downregulation of BET protein levels | NSCLC | apoptosis, inhibition of protective autophagy |
|
| JQ1, AZD5153, NHWD870 | Rapamycin, Everolimus | block of TSC2-mTOR-p70S6K1-BAD pathway | SCLC | intrinsic apoptosis |
|
| JQ1 | ACY-1215 | upregulation of MHC II on tumor cells relies on NK cells | SCLC | apoptosis |
|
| JQ1, AZD5153 | AZD1775 | inhibition of NHEJ activity, enhancement of DNA damage, promotion of mitotic entry and mitotic catastrophe | NSCLC | synergistic cytotoxicity, apoptosis, suppression of proliferation |
|
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCorporate Governance and Law · Accounting and Organizational Management
1 Introduction
Lung cancer remains one of the most prevalent and severe tumors, although the mortality risk continues to decline thanks to improvements in early identification and treatment strategies (Siegel et al., 2022). Of all lung cancer histologic subtypes, the most common (85%) is non-small cell lung cancer (NSCLC), with the remaining 10%–15% being small cell lung cancer (SCLC) and other rare cancers (Siegel et al., 2020). Frustratingly, with a 5-year survival rate of just seven percent, SCLC is the most aggressive subtype of lung cancer (Frese et al., 2021). With the advance of targeted medicines and immunotherapies, the long-term prognosis of lung cancer has dramatically improved; however, it is still less than satisfactory for most advanced lung cancers (Wang et al., 2021).
Genetic mutations have been extensively studied in lung cancer, which change coding sequences (e.g., sequences directly coding for oncoproteins) and non-coding sequences (e.g., promoters and enhancers), directly or indirectly favoring clonal proliferation in cancer cells (Lengauer et al., 1998). Current targeted therapies for lung cancer are also mainly based on genetic mutation. However, disturbances in epigenetic regulation can also lead to the occurrence and metastasis of lung cancer (Dong et al., 2023; Kostyrko et al., 2023). For example, the MYC proto-oncogenes family can broadly affect cell proliferation and apoptosis through complex regulatory networks (Carroll et al., 2018). In addition to the copy number increases from genetic mutations, MYC overexpression is also inextricably linked to surrounding regulatory sequences, with a growing number of studies demonstrating that super-enhancers (SEs) act as culprits in this process. Studies have revealed that the mechanism of SE formation involves mutations, and the uniqueness of epigenetic modifications at SEs loci is related to the progression of acute myeloid leukemia (Cao et al., 2022), colorectal cancer (Liu Q. et al., 2022), and esophageal carcinoma (Shi et al., 2022).
Hence, we introduce the structure and function of SEs and summarize the oncogenic mechanism, classifying functions, and potential therapeutic targets and drugs of SEs in lung cancer here.
2 Subsections
2.1 Identification and structural characterization of SEs
In the 1980s, a 72 bp sequence, later called “enhancer,” was identified in the genome of the SV40/hemoglobin β1 recombinant gene, accompanied by the overexpression of the rabbit β globin gene (Banerji et al., 1981). Enhancers are cis-regulatory elements that interact with promoters from different positions and distances to increase the expression of controlled genes (Blackwood and Kadonaga, 1998). Richard A. Young identified larger clusters of enhancers, known as SEs (or stretch enhancers), which drove high transcription levels of key genes and defined cell identity in pluripotent embryonic stem cells (ESCs) (Whyte et al., 2013). Researchers have created some online repositories such as dbSUPER (Khan and Zhang, 2016) (https://asntech.org/dbsuper/), SEdb (Jiang et al., 2019; Wang Y. et al., 2023) (https://bio.liclab.net/sedb/), SEanalysis (Qian et al., 2019) (https://bio.liclab.net/SEanalysis/), and SEA (Wei et al., 2016; Chen et al., 2020) (http://sea.edbc.org/) to share SEs (and related genes) and their genetic and epigenetic annotations. The constituent enhancers of SEs are with multiple overlapping enrichments of H3K4me1 and H3K27ac of active enhancers (AEs) marks (Yoo et al., 2019; Barral and Déjardin, 2023) (Figure 1). The Rank Ordering of Super Enhancers (ROSE) algorithm is the most commonly used method to identify SEs (Lovén et al., 2013). The algorithm ranks H3K27ac chromatin immunoprecipitation sequencing (ChIP-Seq) signals and defines enhancers above the inflection point (slope >1) as SEs. However, there is still no consensus on an arbitrary cutoff of enrichment level within certain proximity for considering whether an active enhancer is a super-enhancer. As ChIP-seq has advanced, more and more SEs have been discovered to be involved in carcinogenesis, tumor immune evasion (Xu et al., 2019), and various autoimmune disorders (Parker et al., 2013; Vahedi et al., 2015; Yamagata et al., 2022). It is worth mentioning that other techniques, including 4C-seq (Jiang et al., 2020), Hi-C (Huang J. et al., 2018), ChIA-PET (Dowen et al., 2014), ChIP-STARR-seq (Barakat et al., 2018), GRO-seq (Hah et al., 2015), DNase-seq (Kang et al., 2021), and ATAC-seq (Adam et al., 2020), have also been employed to detect SEs in recent years. Lately, Jialiang Huang’s team refined the hierarchical structure of SEs into hub enhancers and non-hub enhancers by hierarchical scoring (Huang J. et al., 2018). Hub enhancers outperform non-hub enhancers in the interaction with regulatory factors and effects on activating target gene transcription.
General definition of SEs. SEs characterized by multiple overlapping enrichments (peaks) of H3K4me1 and H3K27ac are usually composed of several AEs, but there is no consensus on the cutoff of peaks. Multiple coactivators cluster around SEs in a phase-separated manner to activate pro-transcriptional functions. TFs: Transcription factors; AEs: Active enhancers; SEs: Super-enhancers.
As a member of the bromodomain and extra-terminal domain (BET) family and closely associated with SE-related oncogenic transcription, bromodomain-containing protein 4 (BRD4) binds to acetylated modified histones and recruits additional proteins (such as TEFb and Mediator) to indirectly participate in the initiation of transcription and the control of transcriptional elongation (Yang et al., 2005). BET inhibitors (BETi) separate BET proteins from chromatin by competitively binding with histone-acetylated lysines of the BET bromodomain. SEs are highly sensitive to BETi (Fontanals-Cirera et al., 2017). JQ1 is the first reported BETi (Filippakopoulos et al., 2010) and has been widely used for SE identification (Das et al., 2023; Zheng et al., 2024). JQ1 disrupts the transcriptional activity of the master regulators ETS2, HNF4A, and JUNB, suppresses the TGF-induced EMT status, and decreases tumor cell migration and invasion in NSCLC cells (Chang et al., 2016). JQ1, in combination with NSD2 knockdown, inhibits the activation of RAS-driven transcriptional programs in lung cancer cells (García-Carpizo et al., 2016). JQ1 downregulates the expression of HK2, whose promoter is specifically SEs hijacked in a dose-dependent manner in LUAD (Song et al., 2022). In addition, JQ1 exhibits this dose-dependent pharmacological profile in acute myeloid leukemia (Jang et al., 2017), uterine carcinoma (Bonazzoli et al., 2018), and diffuse large B-cell lymphoma (Trabucco et al., 2015).
2.2 Transcription factors (TFs) and core TF involved in coding SE-associated genes
Chromatin accessibility allows physical contact between cis-acting elements and trans-acting factors to regulate transcription processes (Klemm et al., 2019). Breast cancer cells undergoing SE reprogramming produced the accessible chromatin structure and had a higher propensity for lung metastasis (Li K. et al., 2019). TFs are proteins that bind to specific DNA sequences (Spitz and Furlong, 2012). If abnormally enriched at oncogene promoters or enhancers, however, they can result in gene overexpression and carcinogenesis (You et al., 2018). SE-driven genes encode core TFs, which can bind to their SEs or others and interact with other core TFs to regulate the expression of additional genes and form cell- or tissue-specific core regulatory circuits (CRC) (Saint-André et al., 2016; Feng C. et al., 2023). For example, the three specifically SE-activated core TFs ELF3, EHF, and TGIF1 constituted the major component of a CRC with an interdependent pattern that tightly regulated cell migration and invasion in lung adenocarcinoma (LUAD) (Zhang T. et al., 2020).
2.3 Essential SE-associated transcriptional coactivators
Transcriptional coactivators are a variety of proteins with transcription-related functions. As mentioned above, BRD4 is a crucial part of SEs and plays an essential role in the transcriptional activation of oncogenes. Besides, MED1, Mediator, and CBP/p300 with acetyltransferase activity bind to TFs to assist in the assembly of the transcription preinitiation complex and stabilize the transcriptional activity of RNA polymerase II (Ogryzko et al., 1996; Mao et al., 2019; Rengachari et al., 2021). Benjamin R Sabari’s team proposed that transcriptional coactivators enhance transcriptional activity by forming droplets around SEs through a phase separation process (Sabari et al., 2018). However, whether SE-dependent transcriptional enhancement depends on this mechanism is still debatable (Trojanowski et al., 2022).
Overall, the effects of SEs on transcriptional activation are more forceful than that of typical enhancers (TEs) due to their higher region spanning, higher histone modifications, TFs, and RNA polymerase II enrichment, and more binding sites for transcriptional coactivators (Hnisz et al., 2013; Whyte et al., 2013).
2.4 Epigenetic modification associated with SEs
Two key epigenetic regulatory mechanisms involved in SEs are histone modifications (mostly methylation and acetylation) and DNA methylation. Modified histones in the presence of catalytic enzymes (histone lysine methyltransferases or histone acetyltransferases, etc.) can alter chromatin accessibility, affect the assembly of transcriptional complexes, and enhance or reduce the interaction of TFs to gene structures, resulting in altered levels of transcription and translation of downstream genes, which can trigger the development of cancer, autoimmune diseases, and even chronic diseases (Arrowsmith et al., 2012; Fatma et al., 2022; Yang J. et al., 2023). For instance, in a mouse model of LUAD, the deletion of KMT2D (a histone methyltransferase) caused decreased global levels of H3K4me1 and H3K27ac of SEs, leading to the suppression of SE-associated antioncogene Per2 (an antioncogene that inhibits glycolysis) and the activation of glycolytic processes, promoting the growth of cancer (Alam et al., 2020). Another study found that overexpression of NSD2, a histone methyltransferase, resulted in H3K36me2 spreading to the intergenic region of the low H3K27me3 region to exert pro-transcriptional effects (García-Carpizo et al., 2016). That is an example of long-range epigenetic remodeling. It is generally accepted that hypermethylation of CpG islands in the promoter region usually represses gene expression, whereas hypermethylation of gene bodies usually shows the opposite effects (Yang et al., 2014). Interestingly, a study found that highly expressed genes covered by SEs had lower levels of gene body methylation, which resulted in a significant negative correlation between gene body methylation and expression, contrary to popular belief (Pongor et al., 2022). These findings imply an intrinsic link between DNA modification, histone modification, and SEs that jointly influence gene expression.
2.5 SEs formation (or translocation) caused by genetic variation in lung cancer
Several studies have elucidated that the formation (or translocation) of SEs is dependent on genetic variants such as single nucleotide variants (SNVs), insertion deletions (indels), and chromosomal structural variants (SVs) (Mansour et al., 2014; Xing et al., 2019; Wang J. et al., 2023). However, the current research discovered only gene fusion or rearrangement was responsible for SEs’ formation in different tissue subtypes of lung cancer. A study found that focal amplified SEs in LUAD acted on the MYC promoter to spark off transcriptional overactivation (Zhang et al., 2016). And it subsequently demonstrated that the mutations arose from a tandem duplication of a non-coding sequence. In LUAD patients, ALK or ROS1 rearrangements triggered SEs hijacking upstream of EML4 and SLC34A2, driving expression of the new oncogenic fusion genes ALK-EML4 and ROS1-SLC34A2, respectively (Yuan et al., 2021). NUT Carcinoma of the Lung is a rare lung cancer subtype of squamous origin that is extremely aggressive and highly associated with NUT gene fusions (Travis et al., 2015). The fusion gene BRD4/3-NUT co-localized with p300, and the corresponding hyperacetylated region showed a similar SE hijacking function to increase the pro-transcriptional effects of BRD4 (Eagen and French, 2021). Adenoid cystic carcinoma (ACC) originates from the secretory glands and is usually molecularly labeled by the MYB-NFIB fusion oncogene (Persson et al., 2009). MYB rearrangements are widely observed in primary pulmonary adenoid cystic carcinoma (PACC) (Roden et al., 2015). A self-regulatory feedback loop was generated when gene rearrangements (e.g., MYB -NFIB, MYB-TGFBR3, MYB-RAD51B gene fusion) occurring in ACC caused SE translocations and overexpression of MYB proteins (Drier et al., 2016).
2.6 Carcinogenic roles and regulatory mechanisms of SEs in lung cancers
Previous studies on SE-associated activation of oncogenes and signaling pathways have mainly focused on LUAD, as shown in Figure 2. A study showed that focal amplification of the SEs 3′to MYC caused increased expression of MYC and other MYC-target genes (Zhang et al., 2016). The master transcription factor ASCL1 regulates the expression of multiple SE-associated oncogenes, such as RET and MYCL (Miyashita et al., 2018). Nuclear transcription factor estrogen-related receptor alpha (ERRα) coordinates with BRD4 to drive the expression of SE-associated glycolytic gene HK2, leading to enhanced glycolysis progress and growth of malignant cells (Song et al., 2022). Specific SE-driven PADI gene families associated with tumor proliferation, invasion, and colony formation were identified as highly expressed in Osimertinib-resistant LUAD cells (Li H. et al., 2022). SE-associated CENPO was highly expressed in LUAD and positively correlated with the tumor stage, which could promote carcinogenesis and progression by regulating the cell cycle and tumor immune microenvironment (Shi et al., 2023).
Oncogenic mechanisms of SEs in LUAD. Different regulatory factors can directly (binding to SE motifs) or indirectly (recruiting more activators) lead to the overexpression of SE-related genes and the progression (proliferation, invasion, migration) of LUAD. This process may involve the activation of self-regulatory circuits. LUAD, Lung adenocarcinoma.
As we all know, long noncoding RNAs (LncRNAs) are a class of RNAs longer than 200 nt without encoding proteins. Super-enhancer lncRNAs (SE-lncRNAs) are transcribed from super-enhancer genomic regions, which activate the neighboring genes or trans-activate the distant genes through several mechanisms such as transcription factor trapping, promotion of chromatin looping, histone modification, recruitment of phosphorylating RNA polymerase II (Pol II), and isolation of the transcriptional repressor (Schaukowitch et al., 2014; Alvarez-Dominguez et al., 2017; Soibam, 2017; Lee et al., 2020). FOXA1, a driver of multiple cancers (Martínez-Jiménez et al., 2020), can maintain malignant cell survival and promote progression by sensitizing the TGF-β pathway (Wei et al., 2023), activating glycolysis (Xie et al., 2022), and inhibiting autophagy (Li J. et al., 2022). FOXA1 was reported to stimulate LncRNA DSCAM-AS1 at its SEs region (Zhang Y. et al., 2020). DSCAM-AS1, in turn, positively activates FOXA1 expression by interacting with Y box binding protein 1 (YBX1, a DNA/RNA binding protein). Silencing of DSCAM-AS1 inhibits FOXA1 expression and cell growth. Epithelial-mesenchymal transition (EMT), a process by which epithelial cells acquire a mesenchymal cell phenotype and the ability to migrate and invade, is regulated by a series of TFs (Akhmetkaliyev et al., 2023). As a mediator between CDK1 and PRDX1, SE-associated lncRNA LINC00880, activated by TFs FOXP3, leads to phosphorylation of PRDX1 and dysregulation of the PTEN/AKT pathway and metastasis of LUAD (Feng Y. et al., 2023). However, as the LUAD stage progressed, the expression of LINC00880 tended to decrease, and over-expression of LINC00880 had a significant negative prognostic value only in patients with pathological stage I. SEs and related TFs such as ETS2, HNF4A, and JUNB control the high expression of some key EMT genes and define the intermediate state of the EMT process in LUAD cells (Chang et al., 2016). Besides, EMT can be mediated by classical TGF-β/SMAD pathway (Zhang et al., 2014). As one of the receptor-regulated SMAD family members, SMAD3 readily occupies the SE site of LINC01977 and drives its expression in a high TGF-β background (Zhang et al., 2022). In response, LINC01977 boosts the interaction between SMAD3 and CBP/P300 to form a SMAD3/CBP/P300 complex targeting and activating the ZEB1 gene, which induces EMT and causes migration, proliferation, and invasion of LUAD, especially in the early stage of LUAD due to high infiltration of TAM2 (inducing a rich TGF-β environment) and elevated expression of SE-related LINC01977 in this stage. Notably, in p53-deficient lung cancers, the presence of the USP7 (a deubiquitinase) maintains the adherence of the SMAD3 to its own SE elements (EN4, EN9, and EN10), thereby maintaining a positive autoregulatory loop (Huang et al., 2021). However, due to the absence of the p53/MDM2 pathway, USP7 acts as a cancer suppressor. A study showed that TP63-mediated SE-driven LINC01503 was overexpressed and oncogenic in esophageal squamous carcinoma (Xie et al., 2018). Overexpression of LINC01503 was also observed in NSCLC cells and associated with proliferation, migration, and invasion, although it is unclear whether this process is SE-dependent (Shen et al., 2020). Another analysis found that SE-associated lncRNA AC074117.1 was significantly expressed and served as an independent prognostic marker in LUAD (Li et al., 2021).
2.7 Differential landscapes of SEs between LUAD and normal tissues
Several studies comparing LUAD cell lines (or cancerous tissues from patients) with normal tissues have revealed marked differences in SEs landscapes. Enrichment analysis of the respective SE-target genes may help to explain normal physiological processes and tumor carcinogenesis and progression to some extent. LUAD-specific SE-regulated genes are involved in the “lung cancer” disease class and series of oncogenic pathways and are strongly associated with transcriptional disorder. In contrast, normal tissue-specific SE-regulated genes are prominent in fundamental cellular structures and activities (Zhang T. et al., 2020; Zhou et al., 2020; Yuan et al., 2021). Interestingly, from one study, only GO terms (like cell-cell adhesion) with significantly lower SEs counts were observed in cancer cells, which suggested that downregulation of “normal” genes that negatively regulated carcinogenesis could also lead to cancers even when cancer-specific SEs “did not work” (Li X. et al., 2019). A bioinformatics study revealed that cancer-specific H3K27Ac peak had a higher percentage of overlap with accessible chromatin regions to favor transcription than H3K27Ac peak of normal-specific (Jiang et al., 2022).
In conclusion, cancer-specific SEs are more susceptible to other regulators and play a crucial role in determining cancer progression.
2.8 Tumor heterogeneity in SEs helping in typing of lung cancer
Dentro’s whole genome sequence analysis of 2,658 cancer samples from 38 cancer types from the PCAWG project found that genomic instability allowed tumors to evolve into distinct subclonal populations with different biological behaviors among them (Dentro et al., 2021). Epigenomic diversity and instability are widely observed in heterogeneous subclonal populations of multiple types of cancer, involving differences in chromatin accessibility, methylation levels of gene structure (such as gene bodies, promoters, and enhancers of non-coding sequences), and histone profiles (Ding et al., 2019; Pastore et al., 2019; Cejas et al., 2021; El et al., 2023). The analysis of the H3K27Ac peak can directly clarify the distribution characteristics of SEs and assist in identifying associated tumor heterogeneity with them. Figure 3 illustrates the major regulators of SEs and enrichment pathways of SE-related genes in different subgroups of lung cancer.
Subdividing lung cancer subtypes by SE-associated regulatory factors. Differences in SE-associated regulatory mechanisms give rise to differential enrichment of SE-regulated gene sets, which explains the differences in biological behaviors of respective subpopulations to some extent. C/S, Core/Specific; RFs, Regulatory factors; TFs, Transcription factors; EPs, Enrichment pathways; NSCLC, Non-small cell lung cancer; SCLC, Small cell lung cancer; LUAD, Lung adenocarcinoma. LUSC, Lung squamous cell carcinoma.
2.9 High inter-tumor epigenetic heterogeneity presenting in NSCLC
Based on histone modification profiles, a study divided LUAD tissues into two groups that differed significantly in clinical features (e.g., histopathologic subtypes, lymph node invasion), gene mutation patterns, and prognosis (Yuan et al., 2021). Each had its own set of specific core regulatory factors and networks. Specific SE target genes in the poor prognosis group were enriched in invasion- and metastasis-related pathways (e.g., FOXM1, E2F pathway) and dedifferentiated state. In addition, core regulatory networks might shift at early stages, leading to the transition to a terminal stage. Super-enhancer RNA (SeRNA) is known to reflect SE properties and cell specificity (Chen and Liang, 2020; Tu et al., 2021). The distinct groups based on SeRNA profiles showed significant differences in immune infiltration, glycolysis levels, and prognosis (Song et al., 2022). Specifically, tissues in the worst prognostic group had the most chromosomally unstable regions, highest glucose uptake progression, hypoxia-immunotherapy response scores, the most high-risk solid subtype component, and lymph node metastases tendency. A bioinformatics analysis distinguished two subgroups of LUAD with significantly different somatic mutations, survival outcomes, and sensitivities to chemotherapeutic agents by SEs clustering analysis (Jiang et al., 2022). The transcription factor SOX2 usually cooperates with p63 to determine classical subtypes of squamous lung cancer (Watanabe et al., 2014). However, a highly expressed lineage-specific transcription factor Brn2 (an oncogenic factor associated with neuroendocrine carcinomas) was found to replace p63 in lung squamous cell carcinoma (LUSC) cells and to synergize with SOX2 in defining a new “neural” subtype with higher aggressiveness related to poorer prognosis (Sato et al., 2019).
2.10 Fine staging of SCLC using SEs as a hot research topic presently
Investigators have found that differential expression of the TFs ASCL1, NEUROD1, POU2F3, and YAP1 defined four molecular subtypes of SCLC, respectively, and that these TFs co-localized with SEs in the corresponding SCLC subtypes (Borromeo et al., 2016; Huang YH. et al., 2018; Rudin et al., 2019). Multiple lineage-specific TFs are also present in different subtypes of SCLC, and subtype switching occurs even in the same tumor (Ireland et al., 2020; Gay et al., 2021). Focusing on the SCLC-A subtype, Karine Pozo found that the TFs ASCL1, NKX2-1, and PROX1 were associated with SEs and that their interactions formed a regulatory network to control Notch signaling (Pozo et al., 2021). On this basis, Ranran Kong subdivided SCLC-A isoforms and discovered that collaborative NKX2-1/SOX1 regulation defined a new, unique SCLC-Aα subtype (Kong et al., 2022). A recent study found that the complex network of ASCL1-regulated SE-associated miRNAs influenced the identification of SCLC molecular subtypes (Miyakawa et al., 2022). SCLC subtypes identified by TFs differ in EMT status, immune gene expression, immunotherapy (or chemotherapy) benefit, and drug resistance (Gay et al., 2021; Qu et al., 2022). Therefore, subdividing the subtypes of SCLC step by step is an important research direction, which helps us better understand its pathogenesis and develop therapeutic approaches.
2.11 Potential therapeutic targets and clinical/preclinical trials associated with SEs in lung cancer
In recent years, large-scale, individualized molecular profiling of tumors has led to the discovery of an increasing number of targets and therapeutic options to improve the clinical prognosis of patients (Corces et al., 2018; Wu et al., 2022). Due to an inadequate understanding of etiology and pathogenesis and a lack of identified therapeutic targets, more biomarker-driven trials are required to advance therapeutic technologies (William and Glisson, 2011). Transcription of SE-driven genes is highly sensitive to BRD4 inhibitors since studies reveal BETi preferentially repress transcription of SE-related genes relative to TE-related genes (Lovén et al., 2013). The TFIIH complex subunit cyclin-dependent kinase 7 (CDK7), which controls the cell cycle and initiates transcription by Pol II, has emerged as a brand-new target for treating cancer (Larochelle et al., 2012; Suski et al., 2021). Plus, the catalytic member of the P-TEFb complex, cyclin-dependent kinase 9 (CDK9), promotes the transcriptional elongation of Pol II (Zhou et al., 2012). CDK7 can also phosphorylate to activate CDK9 (Larochelle et al., 2012). Therefore, BET, CDK7, and CDK9 may represent viable therapeutic targets for focusing on SE-dependent transcription. Table 1 demonstrates the clinical trials currently underway or have concluded. Besides, a series of preclinical studies of different drug combinations have laid the groundwork for more novel combination therapeutic regimens to enter clinical trials, as shown in Table 2. We provide a brief overview of the results reported.
2.11.1 BETi
The BET protein family member BRD4 is currently the focus of much research. We previously discussed the significance of the first-generation BRD4 inhibitor-JQ1 in the discovery and targeted therapy of SEs. In addition to JQ1, more and more small-molecule drugs targeting BET have been prepared and widely employed in clinical trials (Filippakopoulos et al., 2010).
Birabresib (OTX015, MK-8628), a small molecule complex, was first used in hematologic malignancies. In LUAD cells, the master transcriptional regulators ELF3, EHF, and TGIF1 are downregulated in response to JQ1 and OTX015, causing disruption of CRC structure and inhibition of cancer cells’ malignant progression (Zhang T. et al., 2020).
A Phase I study was designed to evaluate the safety and efficacy of single-agent Birabresib in a range of advanced solid tumors (Lewin et al., 2018). Encouragingly, of the nine NSCLC patients with ALK rearrangement or KRAS mutation, seven developed stable disease (SD), and two developed progressive disease (PD). Though thrombocytopenia was one of the serious adverse effects, it was reversible and self-limiting. Another phase 1/2 study assessed the effectiveness and safety of incrementally applied single-agent Molibresib (Cousin et al., 2022). Of the twelve patients with evaluable SCLC, three developed SD, and nine developed PD. However, no clinically meaningful response criteria were observed, as many patients discontinued dose escalation or even reduced the dose due to drug toxicity. The most common treatment‐related adverse events were thrombocytopenia, nausea, and decreased appetite.
The novel BRD4-targeting agent CFT-2718 showed potent anti-tumor activity in xenograft mice. The intensity and duration of apoptosis induction and tumor proliferation inhibition by CFT-2718 surpassed that of the CDK kinase inhibitor dinaciclib in SCLC cells (Sun et al., 2021). OTX015 showed significant anti-cancer activity in NSCLC cells with or without EML4-ALK translocation and EGFR, KRAS mutations (Riveiro et al., 2016). Meanwhile, in vivo assays demonstrated that OTX015 significantly inhibited the growth of EML4-ALK (+) NSCLC.
2.11.2 CDK7/9 inhibitors
THZ1 inhibits CDK7 activity in an irreversible covalent binding manner (Kwiatkowski et al., 2014). A study found that both SCLC cell lines and mouse models exhibited high sensitivity to THZ1, as evidenced by considerable tumor proliferation inhibition (Christensen et al., 2014). Genes regulated by master-regulated TFs E2F, NRF1, and CREB are preferentially repressed by THZ1 in SCLC cells, suggesting selective effects of THZ1 on SEs and transcriptional core circuits (Christensen et al., 2014), as in LUAD (Zhang T. et al., 2020), neuroblastoma (Chipumuro et al., 2014), and triple-negative breast cancer (Wang et al., 2015). On the other hand, the low sensitivity and specificity of pan-CDK inhibitors for SCLC demonstrate the significance of covalent binding mechanisms in therapeutic efficacy (Christensen et al., 2014). Since the complex formed by BRD4 binding to CDK9/P-TEFb is localized in the SEs region and stimulates the transcriptional activation of MYC, synergistic inhibition of these two targets can effectively halt the progression of cancer (Lovén et al., 2013; Lu et al., 2015).
A phase 2 study compared the difference in efficacy between dinaciclib (a CDK9 inhibitor) and erlotinib in previously treated NSCLC patients (Stephenson et al., 2014). Unfortunately, no patients in the dinaciclib group showed objective responses (OR). The main adverse effects of intravenous denazanil were myelosuppression and gastrointestinal toxicity.
YPN-005 is a novel and selective CDK7 inhibitor with super anti-cancer activity compared to conventional THZ1. YPN-005 showed powerful antiproliferative effects in chemotherapy-resistant SCLC cells, patient-derived organoids, and xenograft mice (Choi et al., 2021). No significant toxic reactions were observed in the mice model. 21e is a highly effective and selective CDK9 inhibitor. After receiving 21e, the proportion of NSCLC side population cells (which indicate stem cell potential) drastically decreased (Wang et al., 2019). A reduction of the cancer stemness marker Oct4, apoptosis of cancer cells, and inhibition of proliferation were observed in xenograft mice treated with 21e.
2.11.3 Combination therapy
It is evident that existing clinical trials, although affirming the therapeutic feasibility of BETi and CDK kinase inhibitors, still suffer from ineffectiveness, inefficacy, toxicity, and resistance. Single-agent (or even partial combination) treatment in these trials does not meet the desired standard. Future studies may favor multiple combination strategies to enhance therapeutic efficacy and reduce toxicity.
Roniclib is a pan-CDK inhibitor (including both CDK7 and CDK9). A phase I/II study reported the potential feasibility of Roniclib in combination with cisplatin/carboplatin-etoposide for extensive SCLC (Cho et al., 2018). Chemotherapeutic agents did not alter the pharmacokinetics of Roniclib but reduced Roniclib exposure by more than one-third. Although none of the 46 patients receiving different doses achieved a complete response (CR), 35 achieved a partial response (PR), five achieved SD, and two developed PD. The most common adverse reactions were nausea, vomiting, anemia, and decreased neutrophil count. This study reflects the favorable efficacy and tolerability of Roniclib for SCLC in all. However, another phase II study comparing the efficacy and safety of Roniciclib in combination with platinum-based chemotherapy versus chemotherapy alone in patients with extensive SCLC did not give us encouraging results (Reck et al., 2019). Although one patient in the combination therapy group achieved CR, overall, the combination therapy group had a lower objective response rate (ORR) and more treatment-emergent adverse events than the platinum-based chemotherapy group. Common adverse reactions were similar to the previous study (Cho et al., 2018). Nausea was the primary cause of discontinuation in the combined therapy group.
Besides, BET- and CDK-targeted inhibitors demonstrate convincing anti-tumor effects in preclinical models and provide paradigms for clinical trials.
BETi can induce cancer cells toward apoptosis by regulating the balance between pro- and anti-apoptotic proteins. QCA570 is a novel BET degrader that caused the degradation of anti-apoptosis-related proteins Mcl-1 in NSCLC cells (Liu C. et al., 2022). QCA570, in combination with ositinib, induced apoptosis and inhibited the proliferation of ositinib-resistant cells in vivo and in vitro experiments. Another study found that the antitumoral activity of the BET inhibitor ABBV-075 in SCLC was partially dependent on the inhibition of BCL2, an anti-apoptotic protein (Lam et al., 2017). In vivo and in vitro assays confirmed the synergistic antitumor effects of ABBV-075 with the BCL2 inhibitor venetoclax (ABT-199), particularly in tumors with a high level of BCL2 expression. Furthermore, combination treatment with JQ1 and the BCL2 inhibitors ABT-263 damaged the balance of Bim and Bcl-2, resulting in the release of the pro-apoptotic protein BIM in MYCN-amplified SCLC cell lines and exerted growth-inhibitory effects in MYCN-positive xenografts (Wang et al., 2017). Another study similarly found that BET inhibitor I-BET762 combined with the PARP inhibitor talazoparib showed higher antitumoral activity in SCLC cells with elevated MYC expression or amplification of MYCs (Fiorentino et al., 2020). The above research implies that the combined strategy boosts the inhibitory effects of BETi on the MYC-related transcriptional pathway.
BET inhibitor treatment alone may result in compensatory alterations that can lower efficacy or develop drug resistance. Therefore, inhibiting these compensatory changes can improve efficacy. For instance, one investigation discovered that JQ1 alone compensatorily upregulated the BET protein (BRD2,3,4), triggered autophagy, and prevented apoptosis in NSCLC cells (Zhou et al., 2021). JQ1 with the chemotherapeutic agent paclitaxel reversed this upregulation and induced apoptosis. Another study found that BETi upregulate the mTOR upstream kinase RSK3, leading to resistance to apoptosis in SCLC cells (Kumari et al., 2023). The combination of BETi and mTOR inhibitors disrupted mTOR signaling and displayed superior anti-cancer activity than any single drug in vitro (JQ1/rapamycin) and in vivo (NHWD870/everolimus) assays.
A combination of inhibitors targeting epigenetic regulators histone deacetylase (HDAC) and BET exerted synergistic antitumoral effects in pancreatic cancer (He et al., 2020), glioblastoma (Gusyatiner et al., 2021), and medulloblastoma (Kling et al., 2022). In SCLC xenograft mice, JQ1 and the histone deacetylase 6 (HDAC6) inhibitor ACY-1215 combined to exert anti-tumor activity dependent on the presence of NK cells (Liu et al., 2018).
Furthermore, JQ1 enhanced DNA double-strand breaks induced by the WEE1 inhibitor AZD1775 and led to mitotic catastrophe, which synergistically produced cytotoxicity and inhibited NSCLC growth in vitro and in vivo experiments (Takashima et al., 2020).
In conclusion, in both in vivo and in vitro studies, whether for SCLC or NSCLC, combination therapies have achieved greater success than any single agent. Combination therapies are also effective in reducing drug resistance and side effects. Based on in-depth studies of more complex carcinogenic pathways, we suppose that more clinical trials will be undertaken in the future.
2.11.4 Other drug candidates in the pan-cancer field
Furthermore, epigenetic modifiers CBP/p300 and HDAC dynamically regulate H3K27 acetylation levels and are potential drug targets in the pan-cancer field (Wang et al., 2022).
CBP/p300 is a histone acetyltransferase that deposits extensive H3K27ac marks and highly occupies SEs (Wang et al., 2022). A study found that the CBP/p300 inhibitor ICG-001 alone upregulated the expression of SE-related oncogenes in glioblastomas, whereas this deleterious effect disappeared when combined with JQ1 (Wiese et al., 2020). Anti-cancer studies with SE-related CBP/p300 inhibitors have been conducted in hematologic malignant neoplasms such as multiple myeloma, acute myeloid leukemia, and non-Hodgkin’s lymphoma (Lasko et al., 2017), as well as in castration-resistant prostate cancer (Chen et al., 2022). Notably, Zhang T et al. found that the EP300 inhibitor CBP30 inhibited the expression of master TFs in LUAD cell lines (Zhang T. et al., 2020).
Studies have confirmed that HDAC inhibitors (HDACi) effectively inhibit the function of SEs (Nagaraja et al., 2017; Sanchez et al., 2018; Nguyen et al., 2020). HDACi targeting SEs exerted great anti-tumor potential in glioblastoma (Nguyen et al., 2020), rhabdomyosarcoma (Gryder et al., 2019), multiple myeloma (Alaterre et al., 2022), stem-like breast cancer (Caslini et al., 2019), ovarian cancer (Shi et al., 2019; Wu et al., 2023), and esophageal squamous cell carcinoma (Jiang et al., 2020). Numerous preclinical studies have confirmed that HDACi inhibits the proliferation of lung cancer cells and is strongly associated with drug resistance (Huang et al., 2014; Wu et al., 2020; He et al., 2022; Eichner et al., 2023). However, it is not yet clear whether this effect is dependent on SEs. Unfortunately, in clinical trials for lung cancer, HDACi are usually used in synergy with radiotherapy, immunotherapy, or epigenetic therapy, as only modest single-agent activity has been demonstrated (Bartling et al., 2005; Traynor et al., 2009; Costa et al., 2014). In any case, drug trials against more SE targets are warranted and expected to provide additional treatment options for lung cancer patients.
3 Conclusion
Since their discovery, SEs have received attention in various fields, and the specific functions they exert in different tumor types are gradually being revealed. The basic structure and function of SEs are similar in different malignant tumors, and all are closely related to cancer progression and aggressiveness. However, it is still to be demonstrated that there are many differences in the SEs characteristics of different cancer types: 1. SEs might be widespread in almost all cancer MYC regions (Zhou and Parsons, 2023), whereas studies of SE changes in the MYC region in lung cancer are limited (Zhang et al., 2016). 2. Gene rearrangements/fusions underlie the formation of cancer-specific SEs. However, compared to hematologic tumors, where high-frequency gene fusions are present, rearrangements/fusions that form SEs are less frequently observed in lung cancer (especially squamous lung cancer) (Li et al., 2016). 3. SE-lncRNA might be widespread in pan-cancer (Ropri et al., 2021; Yan et al., 2021; Chuang et al., 2022; Yang Z. et al., 2023; Hu et al., 2023)。SE-lncRNA has been intensively studied in lung cancer (Xie et al., 2018; Shen et al., 2020; Zhang et al., 2022), hepatocellular carcinoma (Peng et al., 2019; Li et al., 2023; Su et al., 2023; Yuan et al., 2023), and gliomas (Bian et al., 2021; Yang Z. et al., 2023; Chen et al., 2023) but has been rarely reported in hematologic tumors (Handa et al., 2020). However, SEs still cannot be accurately identified due to the low specificity of the classical ChIP-seq technique and the technical imperfections of the Rose algorithm. In addition, the molecular mechanism of how oncogenic signaling alters super enhancer-promoter interactions is not yet understood. For example, how do oncogenic signals promote the aggregation of phase-separated condensates near SEs and drive transcription? How do these signals reshape the three-dimensional folding of the genome, altering the otherwise insulating relationships between regulatory elements and pinpointing the timing, location, and level of transcription?
Here, we summarize the tissue specificity of SEs, mechanisms of SEs formation, oncogenic effects, therapeutic targets, and clinical trials in lung cancer.
Researchers identified subgroups of lung tumors with distinctly diverse biological traits (such as aggressiveness) and prognoses using the Rose algorithms and unsupervised clustering, indicating that these groupings have unique oncogenic signaling networks. However, to date, SEs formed by point mutations or indels have not been identified in the lung cancer genome. Moreover, it is difficult to determine the sequential relationship between the activation of oncogenic pathways and the formation of SEs. Unfortunately, only a few studies related to SEs were conducted for squamous lung cancer. It is challengeable and valuable for researchers to accurately identify SE-driven oncogenic mutations of squamous cell carcinoma, given the close connection between smoking and squamous cell carcinoma and the large number of non-oncogenic passenger mutations resulting from long-term exposure to tobacco carcinogens. BETi and CDK kinase inhibitors are the predominant epigenetic agents targeting SEs. However, existing clinical trials of these drugs are less encouraging due to lack of specificity, development of resistance, and the inevitability of toxicity. Nonetheless, many studies in preclinical models have confirmed the potential benefits of combination therapies and suggested that drug combinations with multiple different mechanisms are an area to explore in the future. It is worth mentioning that the body’s immunological status and the tumor microenvironment are unquestionably significant factors influencing medication action, even though the precise mechanism is still under investigation. Epigenetic therapies can remodel the immune microenvironment, and their use in conjunction with immunosuppressive agents is currently a hot topic in cancer care (Chou et al., 2020; Luo et al., 2022; Tien et al., 2023). In addition to drug therapy, gene editing techniques (e.g., CRISPR/Cas9) can specifically knock down SEs of interest to prevent activation and cascade amplification of oncogenic transcripts. For instance, the knockdown of e3, one of the key components of SEs in the neighboring non-coding region of MYC, downregulates the expression of MYC and downstream targets and inhibits the clonal growth of LUAD cells (Zhang et al., 2016). Therefore, pinpointing functional components within SEs clusters may provide insight into precision medicine. In conclusion, further research into the oncogenic mechanism of SEs will aid in the development of personalized therapeutic strategies and the long-term survival of lung cancer patients.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adam R. C.Yang H.Ge Y.Infarinato N. R.Gur-Cohen S.Miao Y. (2020). NFI transcription factors provide chromatin access to maintain stem cell identity while preventing unintended lineage fate choices. Nat. Cell Biol. 22 (6), 640–650. 10.1038/s 41556-020-0513-0 32393888 PMC 7367149 · doi ↗ · pubmed ↗
- 2Akhmetkaliyev A.Alibrahim N.Shafiee D.Tulchinsky E. (2023). EMT/MET plasticity in cancer and Go-or-Grow decisions in quiescence: the two sides of the same coin? Mol. Cancer 22 (1), 90. 10.1186/s 12943-023-01793-z 37259089 PMC 10230810 · doi ↗ · pubmed ↗
- 3Alam H.Tang M.Maitituoheti M.Dhar S. S.Kumar M.Han C. Y. (2020). KMT 2D deficiency impairs super-enhancers to confer a glycolytic vulnerability in lung cancer. Cancer Cell 37 (4), 599–617. 10.1016/j.ccell.2020.03.005 32243837 PMC 7178078 · doi ↗ · pubmed ↗
- 4Alaterre E.Ovejero S.Herviou L.de Boussac H.Papadopoulos G.Kulis M. (2022). Comprehensive characterization of the epigenetic landscape in Multiple Myeloma. Theranostics 12 (4), 1715–1729. 10.7150/thno.54453 35198065 PMC 8825580 · doi ↗ · pubmed ↗
- 5Alvarez-Dominguez J. R.Knoll M.Gromatzky A. A.Lodish H. F. (2017). The super-enhancer-derived alnc RNA-EC 7/bloodlinc potentiates red blood cell development in trans. Cell Rep. 19 (12), 2503–2514. 10.1016/j.celrep.2017.05.082 28636939 PMC 6013260 · doi ↗ · pubmed ↗
- 6Arrowsmith C. H.Bountra C.Fish P. V.Lee K.Schapira M. (2012). Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discov. 11 (5), 384–400. 10.1038/nrd 3674 22498752 · doi ↗ · pubmed ↗
- 7Banerji J.Rusconi S.Schaffner W. (1981). Expression of a beta-globin gene is enhanced by remote SV 40 DNA sequences. Cell 27 (2 Pt 1), 299–308. 10.1016/0092-8674(81)90413-x 6277502 · doi ↗ · pubmed ↗
- 8Barakat T. S.Halbritter F.Zhang M.Rendeiro A. F.Perenthaler E.Bock C. (2018). Functional dissection of the enhancer repertoire in human embryonic stem cells. Cell Stem Cell 23 (2), 276–288. 10.1016/j.stem.2018.06.014 30033119 PMC 6084406 · doi ↗ · pubmed ↗